Nhóm dược lý điều trị: Thuốc chống ung thư, kháng thể đơn dòng, mã ATC: L01XC21.
Cơ chế tác dụng
Yếu tố tăng trưởng nội mạc mạch máu người (VEGF) Receptor 2 là tác nhân trung gian chính trong sự hình thành mạch. Ramucirumab là một kháng thể đơn dòng nhắm đích mà cụ thể là gắn vào VEGF Receptor 2 và chặn sự gắn kết của VEGF-A, VEGF-C, và VEGF-D. Nhờ vậy, ramucirumab ức chế sự kích hoạt phối tử của VEGF Receptor 2 và các thành phần tín hiệu sau đó, bao gồm enzym p44/p42 protein kinases hoạt hóa mitogen, trung hòa sự tăng sinh do các phối tử và sự di chuyển của tế bào nội mô của con người.
Tính an toàn và hiệu quả lâm sàng
Ung thư dạ dày:
RAINBOW
RAINBOW, một nghiên cứu mù đôi, ngẫu nhiên trên phạm vi toàn cầu, nghiên cứu Cyramza kết hợp paclitaxel kiểm chứng với giả dược kết hợp paclitaxel, đã được thực hiện trên 665 bệnh nhân ung thư dạ dày tái phát tại chỗ và không thể phẫu thuật hoặc di căn (bao gồm cả ung thư biểu mô tuyến GEJ) sau khi đã được hóa trị bởi phác đồ chứa platinum- và fluoropyrimidine, cùng hoặc không cùng anthracycline. Tiêu chí đánh giá chính là thời gian sống còn toàn bộ (OS) và tiêu chí phụ là thời gian sống bệnh không tiến triển (PFS) và tỷ lệ đáp ứng toàn bộ (ORR). Bệnh nhân được lựa chọn phải có bệnh tiến triển trong quá trình hoặc trong vòng 4 tháng sau liều cuối cùng của điều trị bước một và tình trạng sức khỏe ECOG PS 0-1. Bệnh nhân được chọn ngẫu nhiên theo tỷ lệ 1:1 dùng Cyramza kết hợp paclitaxel (n=330) hoặc giả dược kết hợp paclitaxel (n=335). Sự ngẫu nhiên này là ngẫu nhiên về mặt địa lý, thời gian tiến triển từ lúc bắt đầu điều trị bước một (< 6 tháng so với ≥ 6 tháng) và mức độ bệnh. Cyramza liều 8 mg/kg hoặc giả dược được chỉ định truyền tĩnh mạch mỗi 2 tuần (vào ngày 1 và ngày 15) của chu kỳ 28 ngày. Paclitaxel liều 80 mg/m
2 được chỉ định truyền tĩnh mạch vào ngày 1, 8, và 15 của chu kỳ 28 ngày.
Phần lớn (75%) bệnh nhân được chọn ngẫu nhiên trong nghiên cứu trước đó đã dùng trị liệu phối hợp platinum và fluoropyrimidine không có anthracycline. Phần còn lại (25%) trước đó đã dùng trị liệu phối hợp platinum và fluoropyrimidine có anthracycline. Hai phần ba số bệnh nhân đã từng có bệnh tiến triển trong khi vẫn đang điều trị bước một (66,8%). Nhân khẩu học cơ bản của bệnh nhân và đặc tính bệnh học nhìn chung là cân bằng giữa các nhóm: tuổi trung bình là 61 tuổi; 71% số bệnh nhân là nam; 61% số bệnh nhân là người da trắng; 35% là người Châu Á; Tình trạng sức khỏe ECOG là 0 ở 39% số bệnh nhân, 1 ở 61% số bệnh nhân; 81% số bệnh nhân có bệnh có thể đánh giá được và 79% mắc ung thư dạ dày; 21% có ung thư biểu mô tuyến GEJ. Phần lớn bệnh nhân (76%) có bệnh tiến triển trong vòng 6 tháng từ lúc bắt đầu điều trị bước một. Thời gian điều trị trung vị của những bệnh nhân điều trị bằng Cyramza kết hợp paclitaxel là 19 tuần, và thời gian điều trị trung vị của những bệnh nhân điều trị bằng giả dược kết hợp với paclitaxel là 12 tuần. Cường độ liều trung bình tương đối của Cyramza là 98,6% và của giả dược là 99,6%. Cường độ liều trung bình tương đối của paclitaxel là 87,7% ở nhóm Cyramza kết hợp paclitaxel và 93,2% ở nhóm giả dược kết hợp paclitaxel. Một tỷ lệ phần trăm tương tự số bệnh nhân ngừng điều trị do các tác dụng không mong muốn: 12% số bệnh nhân điều trị bằng Cyramza kết hợp paclitaxel so với 11% số bệnh nhân điều trị bằng giả dược kết hợp với paclitaxel. Liệu pháp điều trị chống ung thư toàn diện sau khi ngừng điều trị đã được chỉ định cho 47,9% bệnh nhân dùng Cyramza kết hợp paclitaxel và 46,0% bệnh nhân dùng giả dược kết hợp paclitaxel.
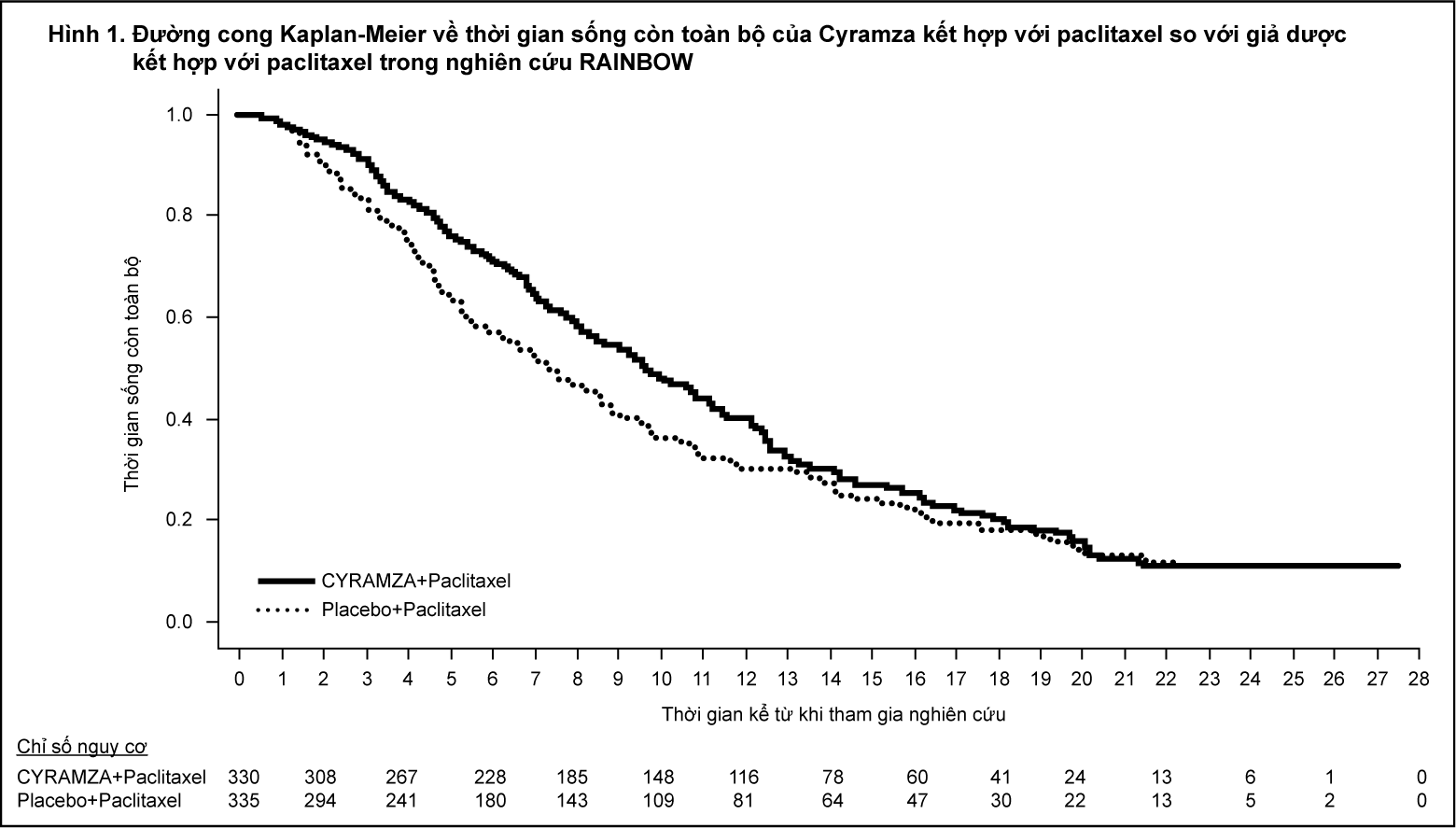
Thời gian sống còn toàn bộ được cải thiện rõ rệt có ý nghĩa thống kê ở những bệnh nhân dùng Cyramza kết hợp paclitaxel so với những bệnh nhân dùng giả dược kết hợp paclitaxel (tỷ số nguy cơ HR 0,807; 95% CI: 0,678 đến 0,962; p=0,0169). Có sự tăng hơn về thời gian sống trung vị 2,3 tháng ở nhóm dùng Cyramza kết hợp với paclitaxel: 9,63 tháng ở nhóm dùng Cyramza kết hợp với paclitaxel và 7,36 tháng ở nhóm dùng giả dược kết hợp paclitaxel.
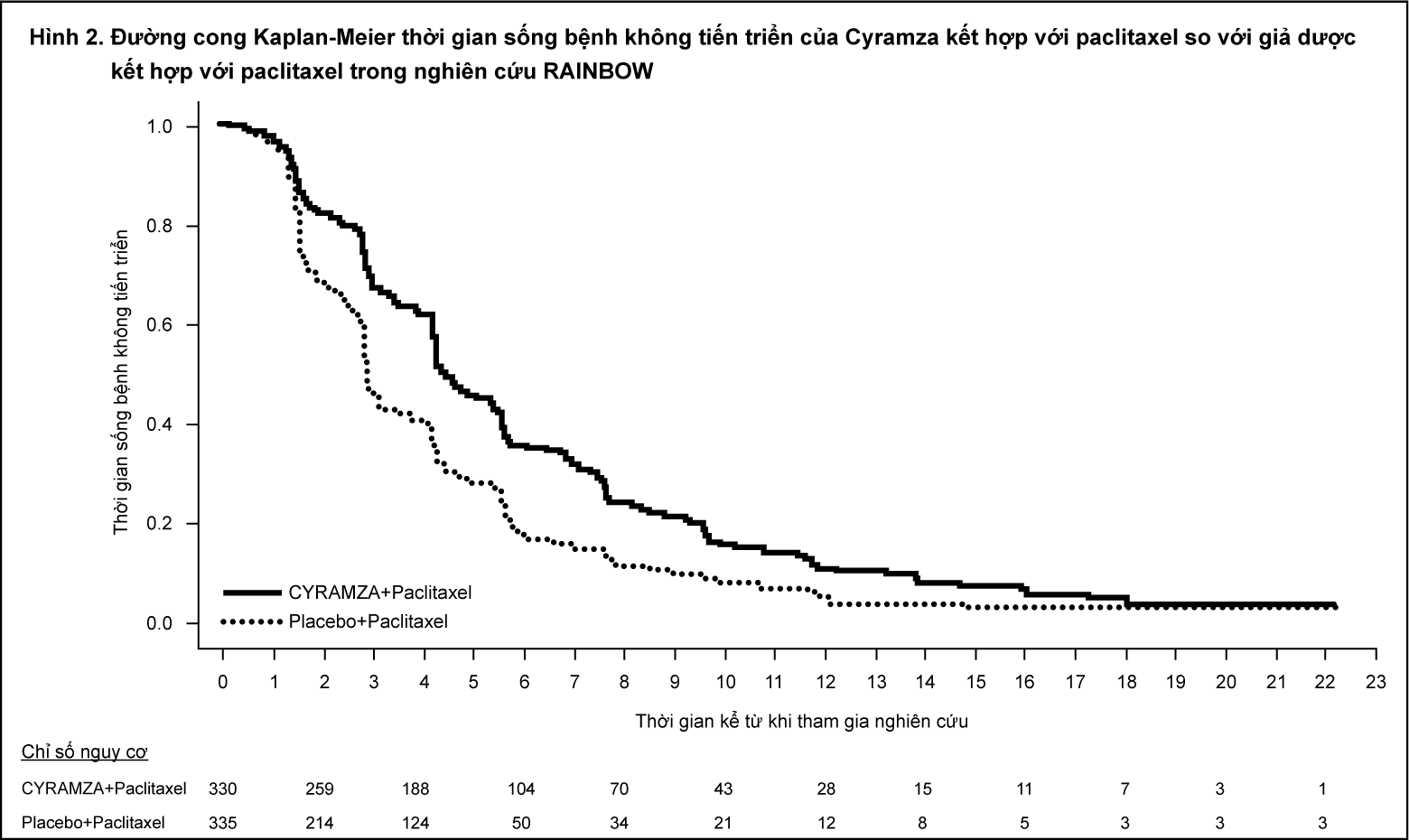
Thời gian sống bệnh không tiến triển (PFS) được cải thiện đáng kể ở nhóm bệnh nhân dùng Cyramza kết hợp paclitaxel so với những bệnh nhân dùng giả dược kết hợp paclitaxel (HR 0,635; 95% CI: 0,536 đến 0,752; p<0,0001). Có sự tăng PFS trung vị nhiều hơn 1,5 tháng ở nhóm Cyramza kết hợp paclitaxel: 4,4 tháng ở nhóm Cyramza kết hợp paclitaxel so với 2,9 tháng ở những bệnh nhân dùng giả dược kết hợp paclitaxel. Tỷ lệ đáp ứng khách quan [ORR (đáp ứng hoàn toàn [CR] + đáp ứng một phần [PR])] được cải thiện đáng kể ở những bệnh nhân dùng Cyramza kết hợp paclitaxel so với những bệnh nhân dùng giả dược kết hợp paclitaxel (tỷ lệ 2,140; 95% CI: 1,499 đến 3,160; p=0,0001). ORR của nhóm Cyramza kết hợp paclitaxel là 27,9% và của nhóm giả dược kết hợp paclitaxel là 16,1%.
Sự cải thiện về OS và PFS được quan sát thấy một cách hằng định ở những phân nhóm định trước về tuổi, giới tính, chủng tộc và trong hầu hết các phân nhóm định trước khác. Các kết quả về hiệu quả được trình bày ở Bảng 1.
- xem Bảng 1, Hình 1 & 2

 REGARD
REGARD
REGARD, một nghiên cứu mù đôi, ngẫu nhiên, đa trung tâm của Cyramza kết hợp với các biện pháp chăm sóc lâm sàng tốt nhất (BSC) so với giả dược kết hợp với BSC, đã được thực hiện trên 355 bệnh nhân ung thư dạ dày tái phát tại chỗ và không thể phẫu thuật hoặc di căn (bao gồm cả ung thư biểu mô tuyến đoạn tiếp nối dạ dày thực quản GEJ) sau khi đã dùng liệu pháp hóa trị trong đó chứa platinum và fluoropyrimidine. Tiêu chí đánh giá chính là thời gian sống còn toàn bộ (OS) và tiêu chí đánh giá phụ là thời gian sống bệnh không tiến triển. Bệnh nhân được lựa chọn phải đang có bệnh tiến triển trong quá trình hoặc trong vòng 4 tháng sau liều cuối cùng của điều trị bước một cho ung thư di căn, hoặc trong quá trình điều trị hỗ trợ hoặc trong vòng 6 tháng sau liều điều trị hỗ trợ cuối cùng, và có ECOG PS 0-1. Để được chọn tham gia nghiên cứu này, bệnh nhân phải có bilirubin toàn phần ≤ 1,5 mg/dL và AST và ALT ≤ 3 lần ULN, hoặc ≤ 5 lần ULN nếu có di căn đến gan.
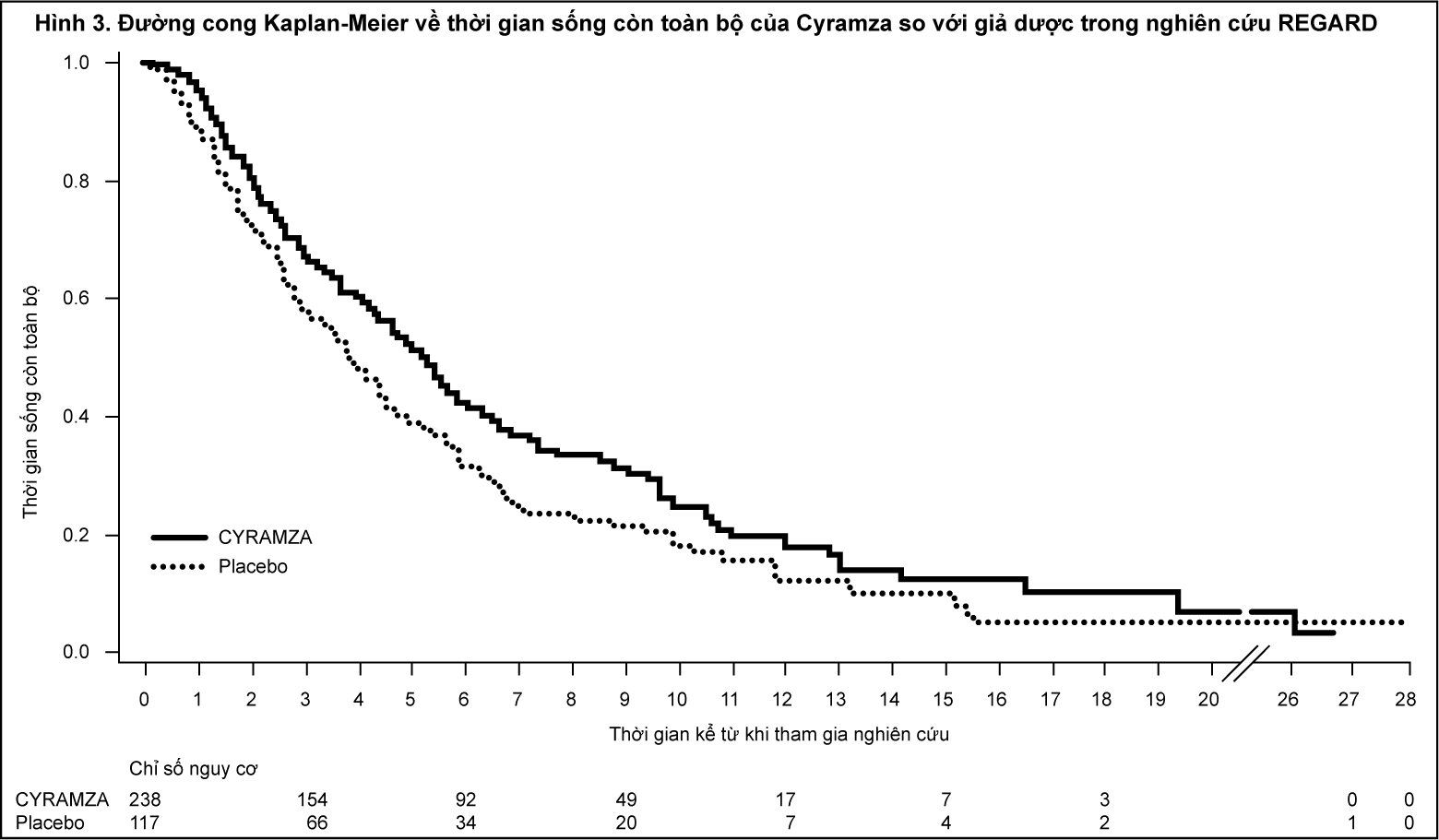
Bệnh nhân được chọn ngẫu nhiên theo tỷ lệ 2:1 truyền tĩnh mạch Cyramza 8 mg/kg (n= 238) hoặc giả dược (n=117) mỗi hai tuần. Sự ngẫu nhiên này được phân tầng bởi mức độ giảm cân trong vòng 3 tháng (≥ 10% so với < 10%), vùng giải phẫu, và vị trí của khối u ban đầu (dạ dày so với đoạn tiếp giáp dạ dày thực quản GEJ). Đặc điểm cơ bản về nhân khẩu và bệnh tật là cân bằng. ECOG PS là 1 ở 72% số bệnh nhân. REGARD không tuyển chọn bệnh nhân nào xơ gan có Child-Pugh B hoặc C. 11% số bệnh nhân điều trị Cyramza và 6% số bệnh nhân điều trị giả dược đã ngừng điều trị do tác dụng không mong muốn. Thời gian sống còn toàn bộ được cải thiện đáng kể ở những bệnh nhân điều trị Cyramza so với những bệnh nhân dùng giả dược (tỷ số nguy cơ [HR] 0,776; 95% CI: 0,603 đến 0,998; p=0,0473), tương đương với việc giảm 22% nguy cơ tử vong và tăng thời gian sống trung vị đến 5,2 tháng khi dùng Cyramza so với 3,8 tháng khi dùng giả dược. Thời gian sống bệnh không tiến triển (PFS) cũng được cải thiện đáng kể ở những bệnh nhân dùng Cyramza so với những bệnh nhân dùng giả dược (HR 0,483; 95% CI: 0,376 đến 0,620; p<0,0001), tương đương với việc giảm được 52% nguy cơ bệnh tiến triển hoặc tử vong và tăng PFS trung vị 2,1 tháng với Cyramza so với 1,3 tháng với giả dược. Các kết quả về hiệu quả của thuốc được trình bày trong Bảng 2.
- xem Bảng 2 & Hình 3

Dựa trên dữ liệu hạn chế từ những bệnh nhân trong nghiên cứu REGARD có ung thư dạ dày HER2 dương tính hoặc ung thư biểu mô tuyến GEJ và những bệnh nhân trước đó đã được điều trị bằng trastuzumab (trong nghiên cứu RAINBOW), người ta cho rằng không chắc Cyramza có tác dụng bất lợi hoặc không có tác dụng đến những bệnh nhân ung thư dạ dày có HER2 dương tính. Phân tích sau đó các phân nhóm không được phân lớp từ những bệnh nhân của nghiên cứu RAINBOW trước đó đã điều trị với trastuzumab (n= 39) cho thấy lợi ích sống còn ở những bệnh nhân này (HR 0,679, 95% CI: 0,327, 1,419) và đã chứng minh được lợi ích của thời gian sống bệnh không tiến triển (PFS) (HR 0,399, 95% CI: 0,194, 0,822).
Ung thư đại trực tràng
RAISE
RAISE là một nghiên cứu mù đôi, ngẫu nhiên, toàn cầu sử dụng Cyramza kết hợp FOLFIRI kiểm chứng với giả dược kết hợp với FOLFIRI, ở những bệnh nhân ung thư đại trực tràng di căn, đã có bệnh tiến triển trong hoặc sau điều trị bước một bằng bevacizumab, oxaliplatin, và một thuốc nhóm fluoropyrimidine. Bệnh nhân phải có ECOG PS 0 hoặc 1 và có bệnh tiến triển trong vòng 6 tháng kể từ liều cuối cùng của trị liệu bước một. Bệnh nhân phải có chức năng gan, thận và khả năng đông máu đạt tiêu chuẩn. Bệnh nhân có tiền sử bệnh di truyền không kiểm soát được hoặc đã từng bị chảy máu hoặc huyết khối, gần đây bị chảy máu nặng (mức độ ≥ 3) hoặc đã từng bị huyết khối động mạch (ATE) trong vòng 12 tháng trước khi được điều trị ngẫu nhiên đều bị loại khỏi nghiên cứu. Bệnh nhân cũng bị loại nếu họ đã từng trải qua bất kỳ: huyết khối động mạch (ATE), tăng huyết áp độ 4, protein niệu độ 3, xuất huyết độ 3-4, hoặc thủng ruột trong quá trình điều trị bước một bằng bevacizumab.
Tổng số 1.072 bệnh nhân được chọn ngẫu nhiên (1:1) dùng hoặc Cyramza (n=536) liều 8 mg/kg hoặc giả dược (n=536) kết hợp với FOLFIRI. Tất cả các thuốc đều được truyền tĩnh mạch. Chế độ liều FOLFIRI như sau: irinotecan 180 mg/m
2 truyền trong hơn 90 phút và folinic acid 400 mg/m
2, dùng đồng thời trong vòng hơn 120 phút; tiếp đó tiêm nhanh 5-fluorouracil (5-FU) 400 mg/m
2 trong hơn 2 đến 4 phút; tiếp tục truyền sau đó 5-FU 2.400 mg/m
2 truyền liên tục trong hơn 46 đến 48 giờ. Chu trình điều trị của cả hai nhóm này được lặp lại mỗi 2 tuần. Bệnh nhân phải ngừng điều trị một hoặc nhiều thành phần thuốc của liệu trình do tác dụng không mong muốn được cho phép tiếp tục điều trị với các thuốc khác cho đến khi bệnh tiến triển hoặc không chịu đựng được độc tính. Tiêu chí đánh giá chính là thời gian sống còn toàn bộ (OS) và tiêu chí phụ là thời gian sống bệnh không tiến triển, tỷ lệ đáp ứng khách quan (ORR) và chất lượng của cuộc sống (QoL) được đo bằng bảng câu hỏi của tổ chức Châu Âu về nghiên cứu và điều trị ung thư (EORTC) QLQ-C30. Sự ngẫu nhiên được phân chia theo vùng địa lý, tình trạng khối u KRAS (đột biến hoặc đơn thuần), và thời gian đến khi tiến triển bệnh (TTP) sau khi bắt đầu điều trị bước một (< 6 tháng so với ≥ 6 tháng).
Đặc điểm nhân khẩu học và tình trạng ban đầu của quần thể bệnh nhân dự định điều trị là tương đương nhau giữa các nhóm điều trị. Tuổi trung bình là 62 tuổi và 40% số bệnh nhân ≥ 65 tuổi; 57% số bệnh nhân là nam; 76% số bệnh nhân là người da trắng; 20% là người Châu Á; 49% số bệnh nhân có ECOG PS 0; 49% bệnh nhân có khối u KRAS đột biến; và 24% số bệnh nhân có thời gian đến khi tiến triển bệnh (TTP) < 6 tháng sau khi bắt đầu điều trị bước một. Liệu pháp điều trị chống ung thư toàn diện sau khi chấm dứt điều trị đã được chỉ định cho 54% bệnh nhân dùng Cyramza kết hợp FOLFIRI và 56% bệnh nhân dùng giả dược kết hợp FOLFIRI.
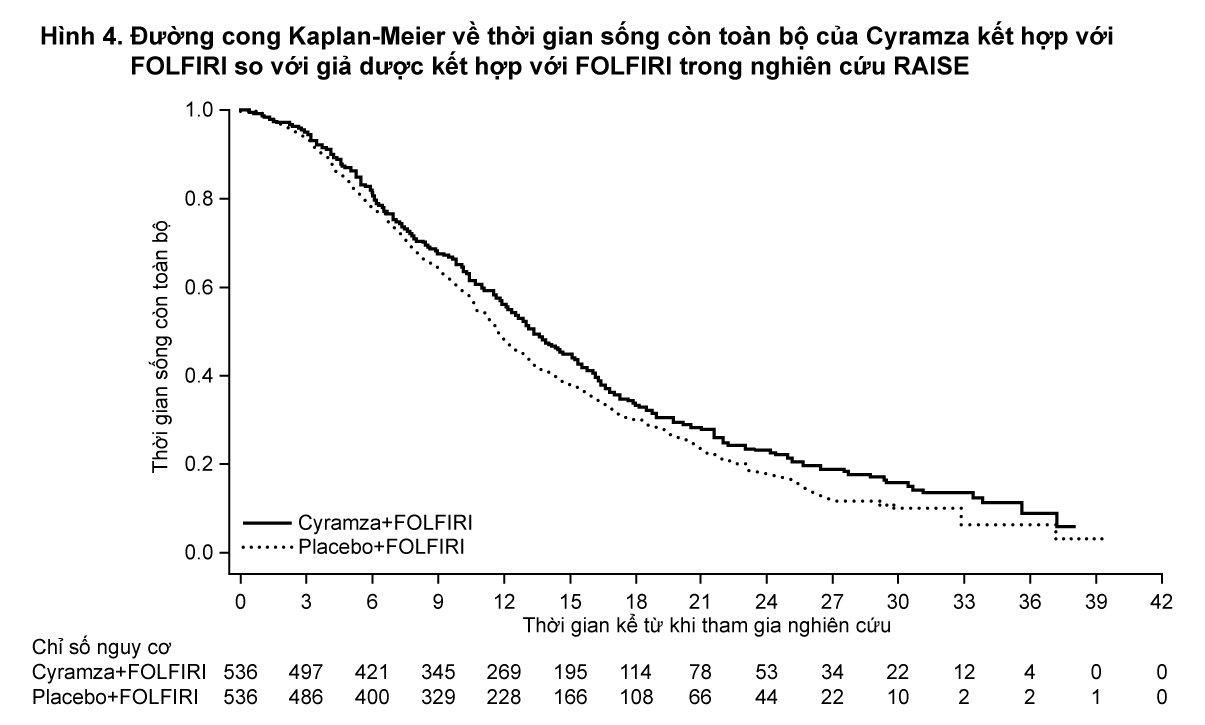
Thời gian sống còn toàn bộ được cải thiện có ý nghĩa thống kê ở những bệnh nhân dùng Cyramza kết hợp FOLFIRI so với những bệnh nhân dùng giả dược kết hợp FOLFIRI (HR 0,844; 95% CI: 0,730 đến 0,976; p=0,0219). Có sự tăng thời gian sống trung vị nhiều hơn 1,6 tháng ở nhóm dùng Cyramza kết hợp FOLFIRI: 13,3 tháng ở nhóm dùng Cyramza kết hợp FOLFIRI và 11,7 tháng ở nhóm dùng giả dược kết hợp FOLFIRI.
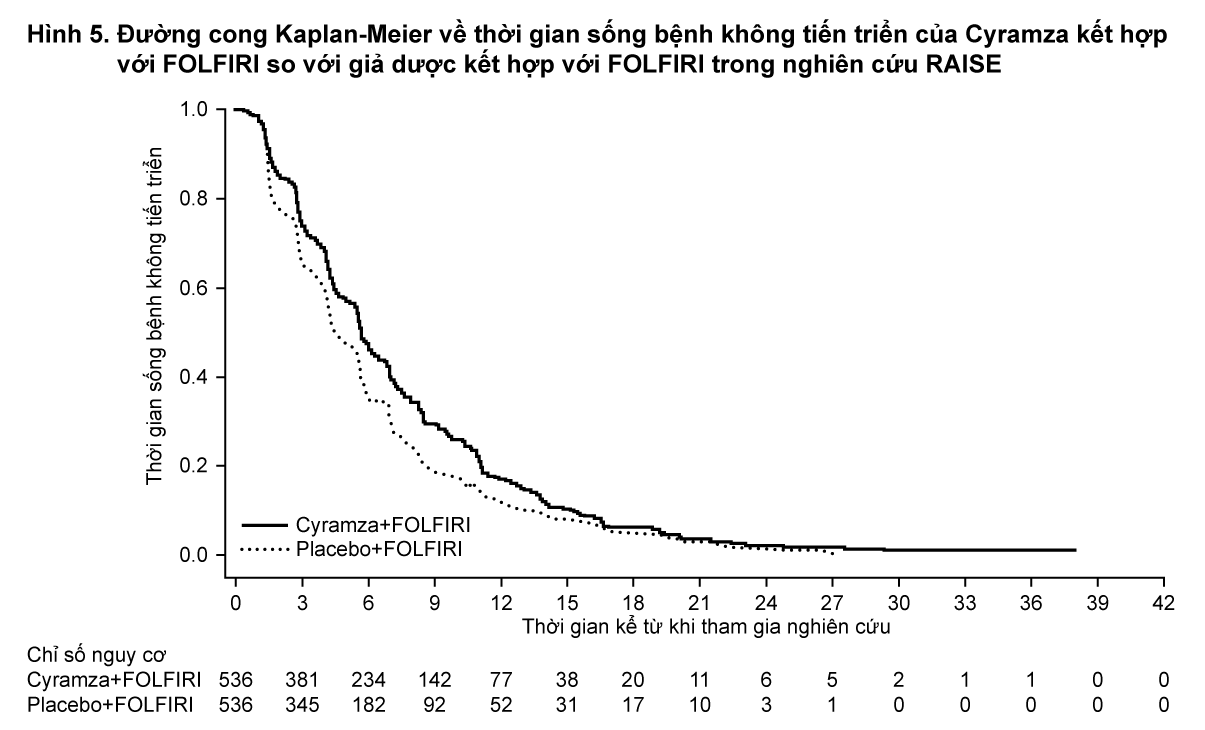
Thời gian sống bệnh không tiến triển (PFS) cũng được cải thiện có ý nghĩa thống kê ở những bệnh nhân dùng Cyramza kết hợp FOLFIRI so với nhóm dùng giả dược kết hợp FOLFIRI (HR 0,793; 95% CI: 0,697 đến 0,903; p=0,0005). Có tăng về trung bình PFS hơn 1,2 tháng ở nhóm dùng Cyramza kết hợp FOLFIRI: 5,7 tháng ở nhóm dùng Cyramza kết hợp FOLFIRI và 4,5 tháng ở nhóm dùng giả dược kết hợp FOLFIRI. Các kết quả về hiệu quả được trình bày trong Bảng 3 và Hình 4 và 5.
Các phân tích tiền phân loại về thời gian sống còn toàn bộ (OS) và thời gian sống bệnh không tiến triển (PFS) bởi các yếu tố phân lớp đã được thực hiện. Tỷ số nguy cơ của OS là 0,82 (95% CI: 0,67 đến 1,0) ở những bệnh nhân có khối u KRAS đơn thuần, và 0,89 (95% CI: 0,73 đến 1,09) ở những bệnh nhân có khối u KRAS đột biến. Đối với những bệnh nhân có thời gian đến khi bệnh tiến triển (TTP) ≥ 6 tháng sau khi bắt đầu điều trị bước một, tỷ số nguy cơ của OS là 0,86 (95% CI: 0,73 đến 1,01), và 0,86 (95% CI: 0,64 đến 1,13) ở những bệnh nhân có TTP < 6 tháng sau khi bắt đầu điều trị bước một. Các phân tích tiền phân loại phân nhóm phụ cho cả PFS và OS theo tuổi (< 65 và ≥ 65 tuổi), giới tính, chủng tộc, ECOG PS (0 hoặc ≥ 1), số cơ quan liên quan, chỉ di căn gan, vị trí của khối u ban đầu (ruột hay trực tràng), nồng độ CEA (< 200 μg/L, ≥ 200 μg/L), tất cả cho thấy hiệu quả điều trị cao hơn ở nhóm điều trị Cyramza kết hợp FOLFIRI so với giả dược kết hợp FOLFIRI. 32 trong số 33 phân tích tiền phân loại phân nhóm về OS, tỷ số nguy cơ < 1,0. Có một phân nhóm có tỷ số nguy cơ (HR) > 1 là những bệnh nhân tiến triển từ lúc bắt đầu điều trị bước một bằng bevacizumab < 3 tháng (HR 1,02 [95% CI: 0,68 đến 1,55]). Phân nhóm phụ này là một nhóm có thể coi là mắc bệnh nặng mà tương đối khó để điều trị bước một. Trong cả hai nhóm điều trị, bệnh nhân đã từng bị giảm bạch cầu trung tính có thời gian OS trung bình dài hơn so với những bệnh nhân chưa từng bị. Thời gian OS trung vị ở những bệnh nhân bị giảm bạch cầu trung tính ở bất cứ độ nào của nhóm dùng ramucirumab (16,1 tháng) đều cao hơn ở so với nhóm dùng giả dược (12,6 tháng). OS trung vị ở những bệnh nhân chưa từng bị giảm bạch cầu là 10,7 tháng ở cả hai nhóm.
- xem Bảng 3, Hình 4 & 5

Tỷ lệ đáp ứng khách quan (ORR) tương tự trong cả hai nhóm điều trị (13,4% so với 12,5%, tương ứng với ramucirumab kết hợp với FOLFIRI so với giả dược kết hợp với FOLFIRI). Tỷ lệ khống chế bệnh (đáp ứng hoàn toàn cộng với đáp ứng một phần cộng với bệnh ổn định) cao hơn ở nhóm những bệnh nhân dùng ramucirumab kết hợp FOLFIRI so với nhóm giả dược kết hợp FOLFIRI (tương ứng là 74,1% so với 68,8%). Về EORTC QLQ-C30, nhóm những bệnh nhân dùng ramucirumab kết hợp FOLFIRI được báo cáo có sự giảm nhẹ QoL so với nhóm giả dược kết hợp FOLFIRI ở hầu hết các chỉ số đo. Đã có các báo cáo về một vài sự khác biệt giữa các nhóm sau tháng đầu tiên điều trị.
Ung thư phổi không tế bào nhỏ (UTPKTBN)
REVEL
REVEL, một nghiên cứu mù đôi, ngẫu nhiên của Cyramza kết hợp với docetaxel so với giả dược kết hợp với docetaxel, đã được thực hiện trên 1.253 bệnh nhân mắc UTPKTBN tiến triển tại chỗ hoặc di căn vảy hoặc không vảy có bệnh tiến triển trong hoặc sau một trị liệu platinum. Tiêu chí chính là thời gian sống còn toàn bộ (OS). Bệnh nhân được chọn ngẫu nhiên với tỷ lệ 1:1 dùng Cyramza kết hợp với docetaxel (n=628) hoặc giả dược kết hợp với docetaxel (n=625). Sự ngẫu nhiên được phân loại bởi vùng địa lý, giới tính, trước duy trì, và ECOG PS. Cyramza liều 10 mg/kg hoặc giả dược và docetaxel liều 75 mg/m
2 được chỉ định truyền tĩnh mạch vào ngày 1 của chu kỳ 21 ngày. Các vùng ở Đông Á được chỉ định liều docetaxel thấp hơn là 60 mg/m
2 mỗi 21 ngày. Bệnh nhân gần đây bị bệnh phổi, tiêu hóa nặng hoặc chảy máu hậu phẫu, có biểu hiện xuất huyết thần kinh trung ương, có khối u ở đường hô hấp chính hoặc mạch máu, bọt khí trong khối u, và tiền sử xuất huyết đáng kể hoặc rối loạn huyết khối không kiểm soát được đều bị loại trừ. Những bệnh nhân có điều trị bất kỳ thuốc chống đông máu nào và/hoặc điều trị bệnh mạn tính bằng các thuốc chống viêm không steroid hoặc các tác nhân kháng tiểu cầu khác hoặc những bệnh nhân di căn thần kinh trung ương/não không ổn định về mặt lâm sàng không được điều trị cũng đều bị loại trừ. Cho phép sử dụng aspirin với liều đến 325 mg/ngày (xem phần
Cảnh báo và thận trọng). Một số lượng hạn chế bệnh nhân không phải da trắng, đặc biệt bệnh nhân da đen (2,6%) được tham gia. Vì thế, kinh nghiệm sử dụng phối hợp ramucirumab và docetaxel ở những bệnh nhân UTPKTBN giai đoạn muộn cũng như những bệnh nhân suy thận, bệnh tim mạch hoặc béo phì còn hạn chế.
Đặc điểm nhân khẩu học và tình trạng bệnh ban đầu của bệnh nhân nhìn chung là tương đương giữa các nhóm điều trị: Tuổi trung bình là 62 tuổi; 67% số bệnh nhân là nam; 82% số bệnh nhân là người da trắng; 13% số bệnh nhân là người Châu Á; 32% số bệnh nhân có ECOG PS 0, 67% số bệnh nhân có ECOG PS 1; 73% số bệnh nhân có mô tế bào không vảy và 26% số bệnh nhân có mô tế bào vảy. Các liệu pháp điều trị phổ biến nhất trước đó là pemetrexed (38%), gemcitabine (25%), taxane (24%), và bevacizumab (14%); 22% số bệnh nhân trước đó đã điều trị duy trì. Thời gian điều trị docetaxel trung vị là 14,1 tuần ở nhóm ramucirumab kết hợp docetaxel (nhận được trung bình 4,0 lần truyền) và 12,0 tuần ở nhóm giả dược kết hợp docetaxel (nhận được trung bình 4,0 lần truyền).
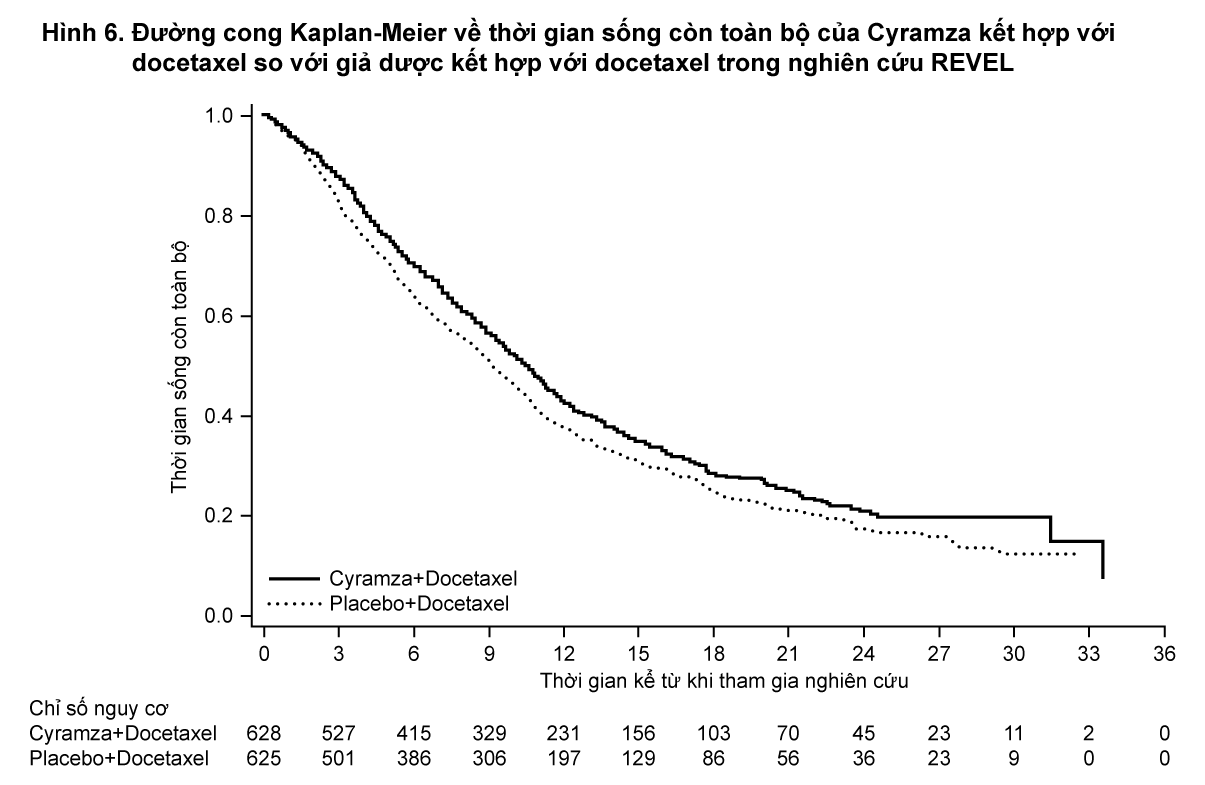
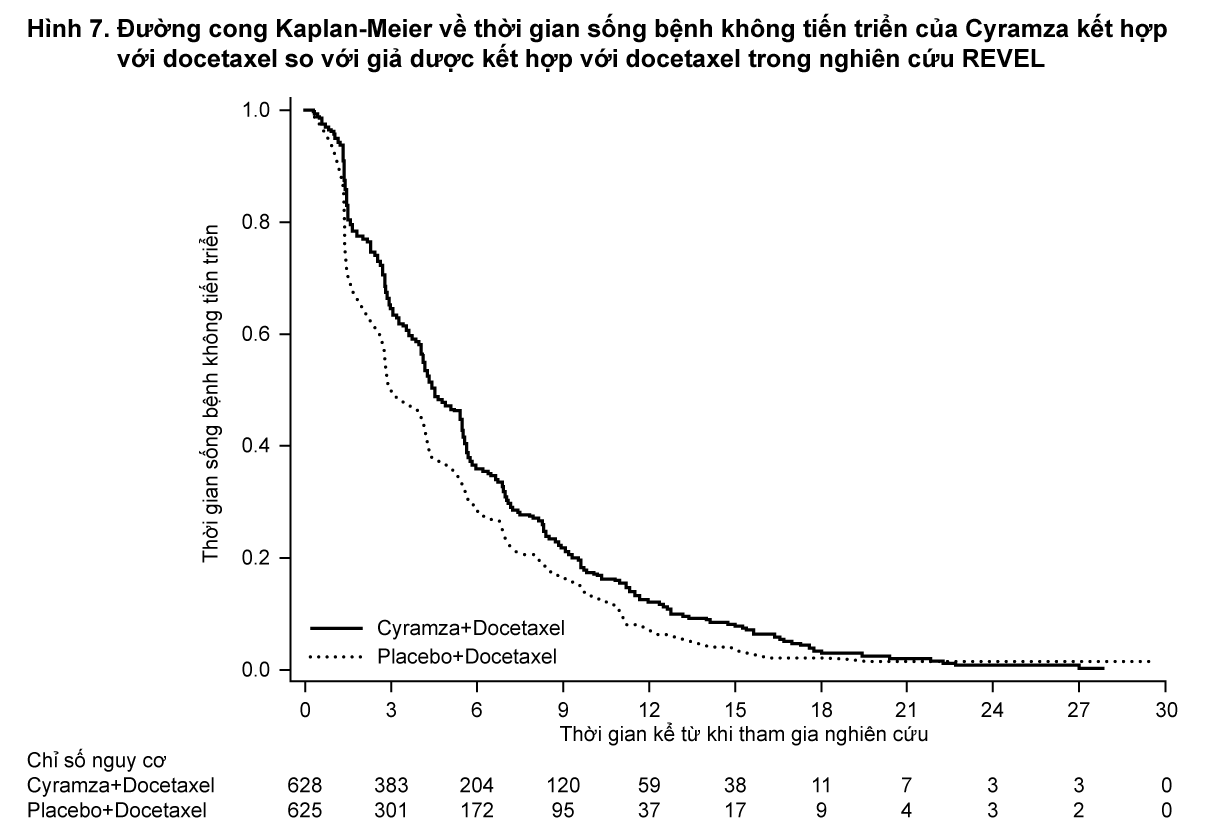
Thời gian sống còn toàn bộ được cải thiện có ý nghĩa thống kê ở những bệnh nhân dùng Cyramza kết hợp docetaxel so với những bệnh nhân dùng giả dược kết hợp docetaxel (HR 0,857; 95% CI: 0,751 đến 0,979; p=0,024). Có sự tăng thời gian sống trung vị nhiều hơn 1,4 tháng ở nhóm dùng Cyramza kết hợp docetaxel: 10,5 tháng ở nhóm dùng Cyramza kết hợp docetaxel và 9,1 tháng ở nhóm dùng giả dược kết hợp docetaxel. Thời gian sống bệnh không tiến triển cũng được cải thiện có ý nghĩa thống kê ở những bệnh nhân dùng Cyramza kết hợp docetaxel so với nhóm dùng giả dược kết hợp docetaxel (HR 0,762; 95% CI: 0,677 đến 0,859; p<0,001). Có tăng về trung vị thời gian sống bệnh không tiến triển (PFS) hơn 1,5 tháng ở nhóm dùng Cyramza kết hợp docetaxel: 4,5 tháng ở nhóm dùng Cyramza kết hợp docetaxel và 3 tháng ở nhóm dùng giả dược kết hợp docetaxel. Tỷ lệ đáp ứng khách quan (ORR) được cải thiện đáng kể ở những bệnh nhân dùng Cyramza kết hợp docetaxel so với giả dược kết hợp với docetaxel (22,9% so với 13,6%, p<0,001). Phân tích QoL chính cho thấy thời gian đến khi suy giảm các điểm số cho tất cả các thang điểm triệu chứng ung thư phổi là tương tự nhau giữa hai nhóm điều trị.
Đã ghi nhận có sự cải thiện về thời gian sống còn toàn bộ (OS) và thời gian sống bệnh không tiến triển (PFS) một cách hằng định (ramucirumab kết hợp với docetaxel so với giả dược kết hợp với docetaxel) ở những phân nhóm quan trọng. Các kết quả OS của các phân nhóm như sau: mô học không vảy (tỷ số nguy cơ (HR) 0,83; 95% CI: 0,71 đến 0,97; OS trung bình [mOS]: 11,1 so với 9,7 tháng) và mô tế bào vảy (HR 0,88; 95% CI: 0,69 đến 1,13; mOS: 9,5 so với 8,2 tháng); bệnh nhân trước đó đã điều trị duy trì (HR 0,69; 95% CI: 0,51 đến 0,93; mOS: 14,4 so với 10,4 tháng); thời gian từ lúc bắt đầu điều trị trước đó < 9 tháng (HR 0,75; 95% CI: 0,64 đến 0,88; mOS: 9,3 so với 7,0 tháng); bệnh nhân < 65 tuổi (HR 0,74, 95% CI: 0,62, 0,87; mOS: 11,3 so với 8,9 tháng). Đã ghi nhận xu hướng ít hiệu quả hơn khi tuổi càng tăng ở những bệnh nhân điều trị ramucirumab kết hợp docetaxel cho điều trị UTPKTBN giai đoạn muộn có sự tiến triển bệnh sau hóa trị liệu bằng platinum (xem phần
Dược lực học). Không ghi nhận thấy sự khác biệt về mặt hiệu quả giữa các nhóm điều trị ở những phân nhóm có bệnh nhân ≥ 65 tuổi (OS HR 1,10, 95% CI: 0,89, 1,36; OS trung bình [mOS]: 9,2 so với 9,3 tháng, xem phần
Cảnh báo và thận trọng), bệnh nhân trước đó đã điều trị bằng taxanes (HR 0,81; 95% CI: 0,62 đến 1,07; mOS 10,8 so với 10,4 tháng) và với những bệnh nhân mà thời gian bắt đầu điều trị trước đó ≥ 9 tháng (HR 0,95; 95% CI: 0,75 đến 1,2; mOS: 13,7 so với 13,3 tháng). Các kết quả về hiệu quả được trình bày trong Bảng 4.
- xem Bảng 4, Hình 6 & 7

Bệnh nhân có điểm số ECOG ≥ 2 bị loại khỏi các nghiên cứu then chốt trong tất cả các chỉ định, vì thế tính an toàn và hiệu quả của Cyramza ở những bệnh nhân này chưa được biết đến.
Tính sinh miễn dịch
Bệnh nhân ở hai nghiên cứu pha 3, RAINBOW và REGARD được kiểm tra ở nhiều thời điểm về các kháng thể kháng thuốc (ADAs). Các mẫu được kiểm tra từ 956 bệnh nhân: 527 bệnh nhân điều trị ramucirumab và 429 bệnh nhân điều trị đối chứng. 11 (2,2%) bệnh nhân điều trị ramucirumab và 2 (0,5%) bệnh nhân điều trị đối chứng có ADAs. Không có bệnh nhân ADAs nào mắc IRR (phản ứng liên quan đến truyền thuốc). Không có bệnh nhân nào có kháng thể trung hòa ramucirumab. Không có đủ dữ liệu để đánh giá tác động của ADAs đến tính hiệu quả hoặc an toàn của ramucirumab.
Nhóm bệnh nhân nhi
Cơ quan quản lý thuốc Châu Âu đã chấp thuận miễn nộp kết quả nghiên cứu Cyramza cho tất cả các phân nhóm bệnh nhân nhi mắc ung thư biểu mô tuyến dạ dày, ung thư biểu mô tuyến đại tràng và trực tràng và ung thư phổi (xem phần
Liều lượng và cách dùng để biết thông tin về việc sử dụng trên bệnh nhân nhi).












 Đăng xuất
Đăng xuất