



 Sign Out
Sign Out
Effect on prostate gland: In a receptor-binding assay using human prostate membrane specimens, silodosin showed a high affinity to the α1A-adrenergic receptor subtype.
Silodosin inhibited noradrenalin-induced contractions of human prostate smooth muscle.
Effect in animals: Effect on lower urinary tract tissue (prostate, urethra, and trigone of the urinary bladder).
Silodosin exhibited a potent antagonistic action against noradrenalin-induced contractions in isolated rabbit prostate, urethra, and trigone of the urinary bladder.
Effect on urethral pressure: In anesthesized male rats, phenylephrine-induced increases in urethral pressure in the region of the prostate were selectively inhibited by silodosin. The inhibitory dose was lower than hypotensive dose.
In anesthetized male dogs, increased urethral pressure in the region of the prostate due to electrical stimulation of the hypogastric nerve was also selectively inhibited by silodosin. The inhibitory dose was lower than the hypotensive dose.
Effect in prostatic hypertrophy model: In a male rat prostatic hypertrophy model prepared by administration of sex hormone, bladder irritation symptoms associated with urinary retention were inhibited.
Mechanism of Action: By blocking the sympathetic nervous system, which mediates the α1A-adrenoreceptor subtype which is distributed in lower urinary tract tissue (prostate, urethra, and trigone of the urinary bladder), silodosin reduces smooth muscle tone of lower urinary tract tissue and inhibits increases in urethral pressure, thereby improving lower urinary tract symptoms associated with benign prostatic hyperplasia.
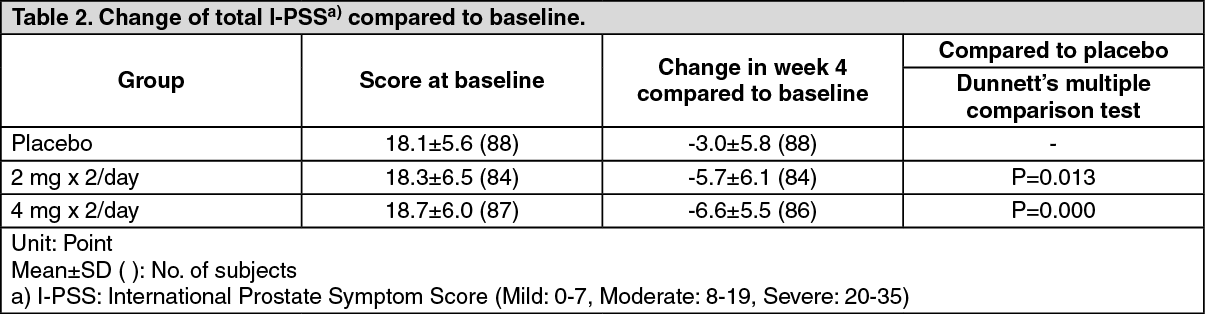
Clinical Studies: Phase II-Double Blind Comparative Study: When silodosin at a dose of 2 or 4 mg, or placebo was administered orally twice daily for 4 weeks to patients with lower urinary tract symptoms associated with benign prostatic hyperplasia, subjective symptoms (total I-PSS) were significantly improved in the 4-mg group compared to the placebo group. (See Table 2.)
 Click on icon to see table/diagram/image
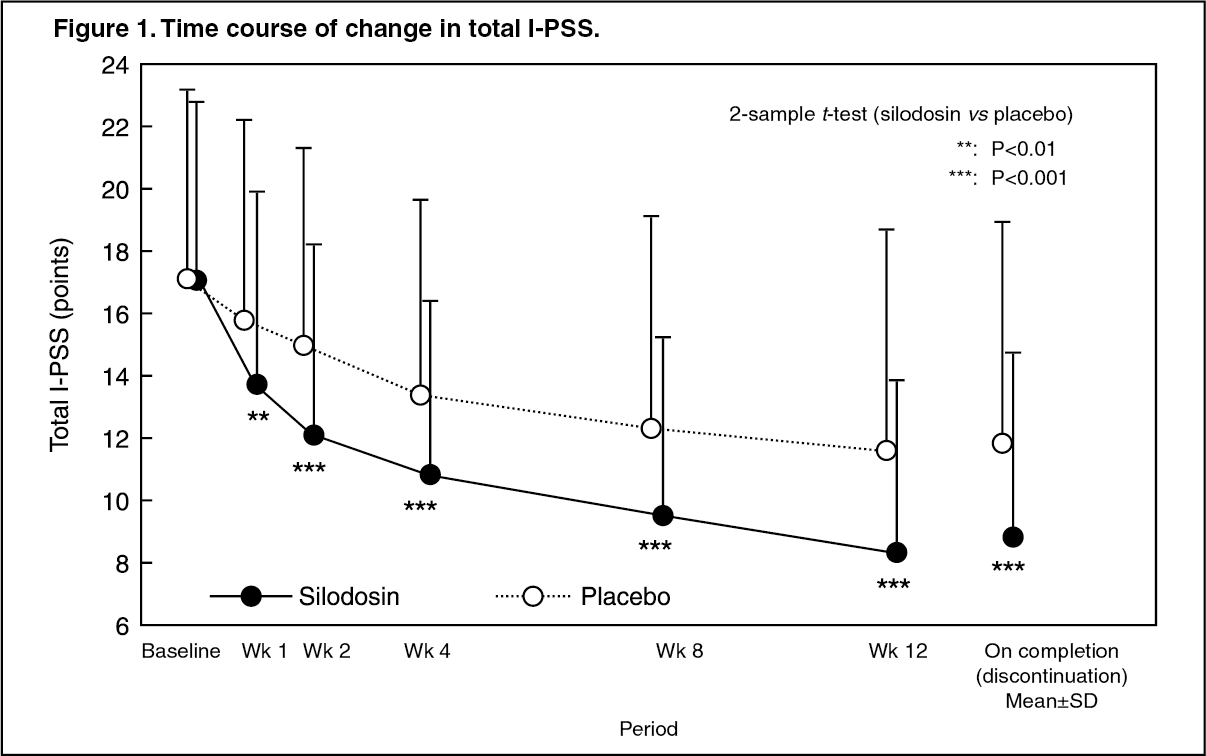
Click on icon to see table/diagram/imagePhase III Double-blind Comparative Study: When silodosin at a dose of 4 mg or placebo was administered orally twice daily for 12 weeks to patients with lower urinary tract symptoms associated with benign prostatic hyperplasia, the total I-PSS in the silodosin and placebo groups on completion of the study showed a decrease of 8.3 and 5.3 points, respectively, compared to baseline (see Figure 1 and Table 3). The percentage (%) of patients in the silodosin and placebo groups whose total I-PSS improved by at least 25% compared to baseline was 76.4% (133/174 patients) and 50.6% (45/89 patients), respectively. The percentage (%) of patients in the silodosin and placebo groups whose symptoms improved to mild (total I-PSS: <8) was 47.7% (83/174 patients) and 31.5% (28/89 patients), respectively.
In the silodosin group, an improvement in subjective symptoms was seen from as early as wk 1 and an improvement effect was also observed in patients whose symptoms were severe. (See Figure 1 & Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image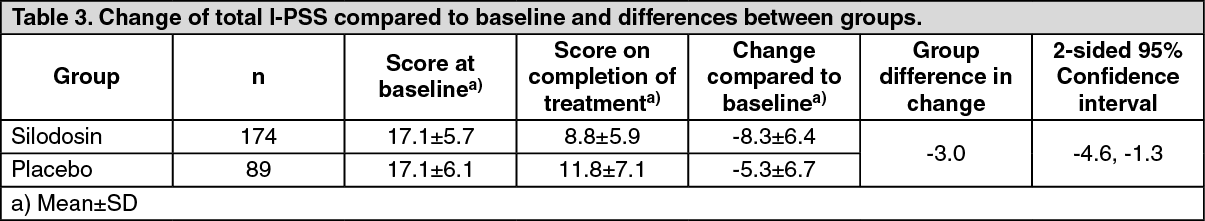 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLong-term Administration Study: In a long-term administration study, silodosin was administered at a dose of 4 mg twice daily for 52 weeks to 364 patients with lower urinary tract symptoms associated with benign prostatic hypertrophy. A continuous improvement effect and drug safety were reported and stable subjective symptoms (total I-PSS) and improvement in maximum urine flow rate were observed.
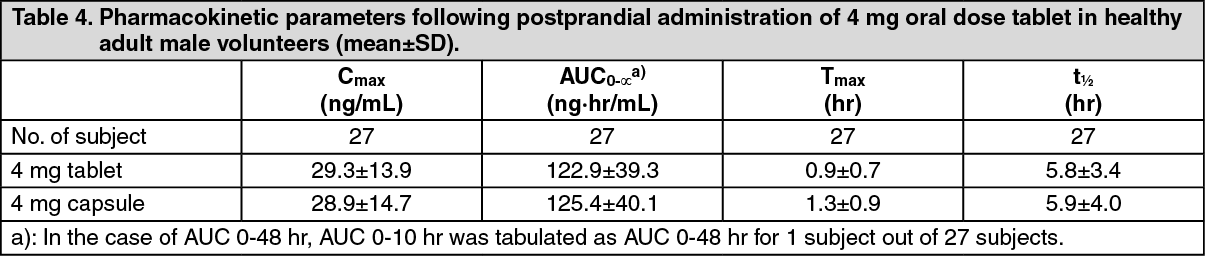
Pharmacokinetics: Absorption and Plasma Concentrations: When a single 4 mg dose of silodosin tablet or capsule was administered orally to 27 healthy adult males, respectively, plasma concentrations and pharmacokinetic parameters of silodosin tablet are shown in Table 4 and Figure 2.
It was demonstrated that the silodosin tablet of 4 mg and capsule of 4 mg are biologically equivalent. (See Table 4 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image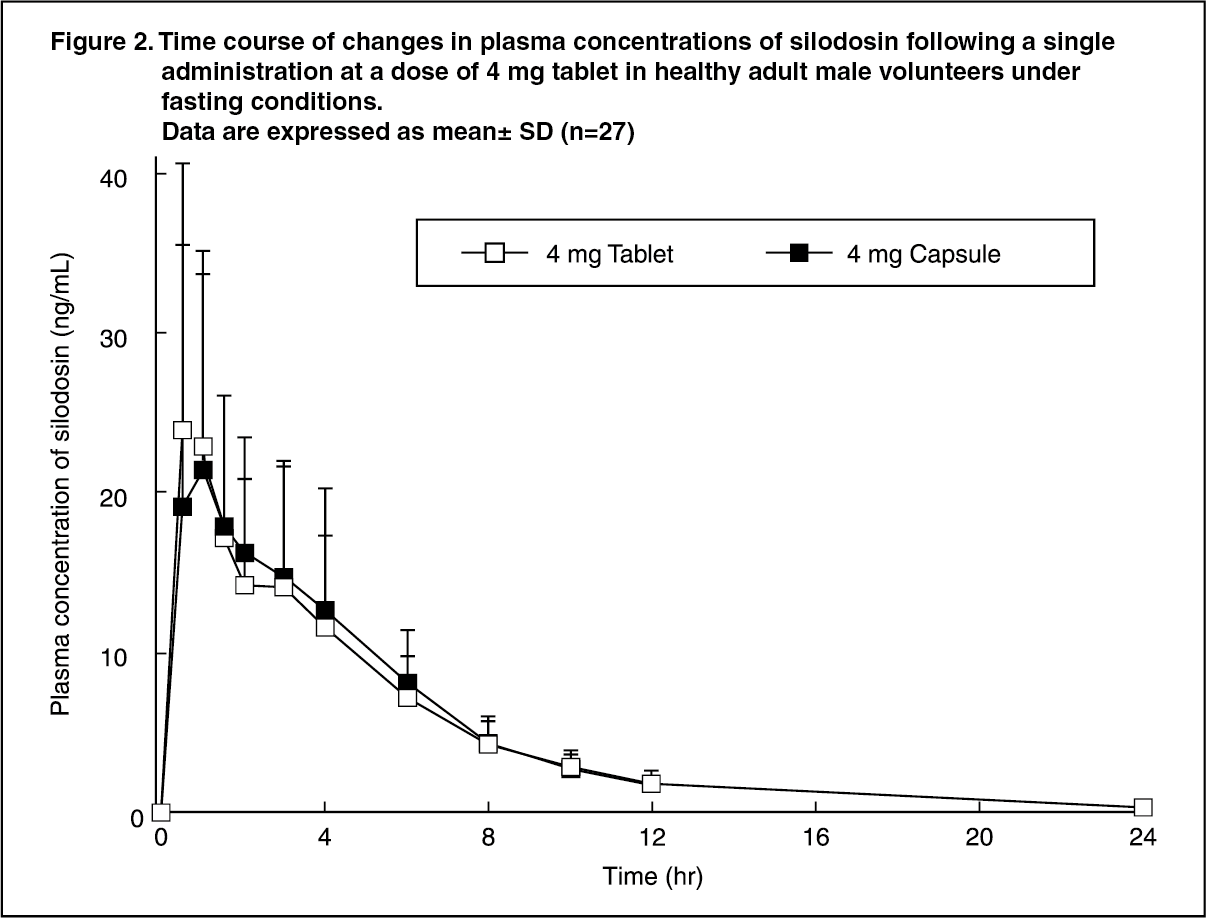 Click on icon to see table/diagram/image
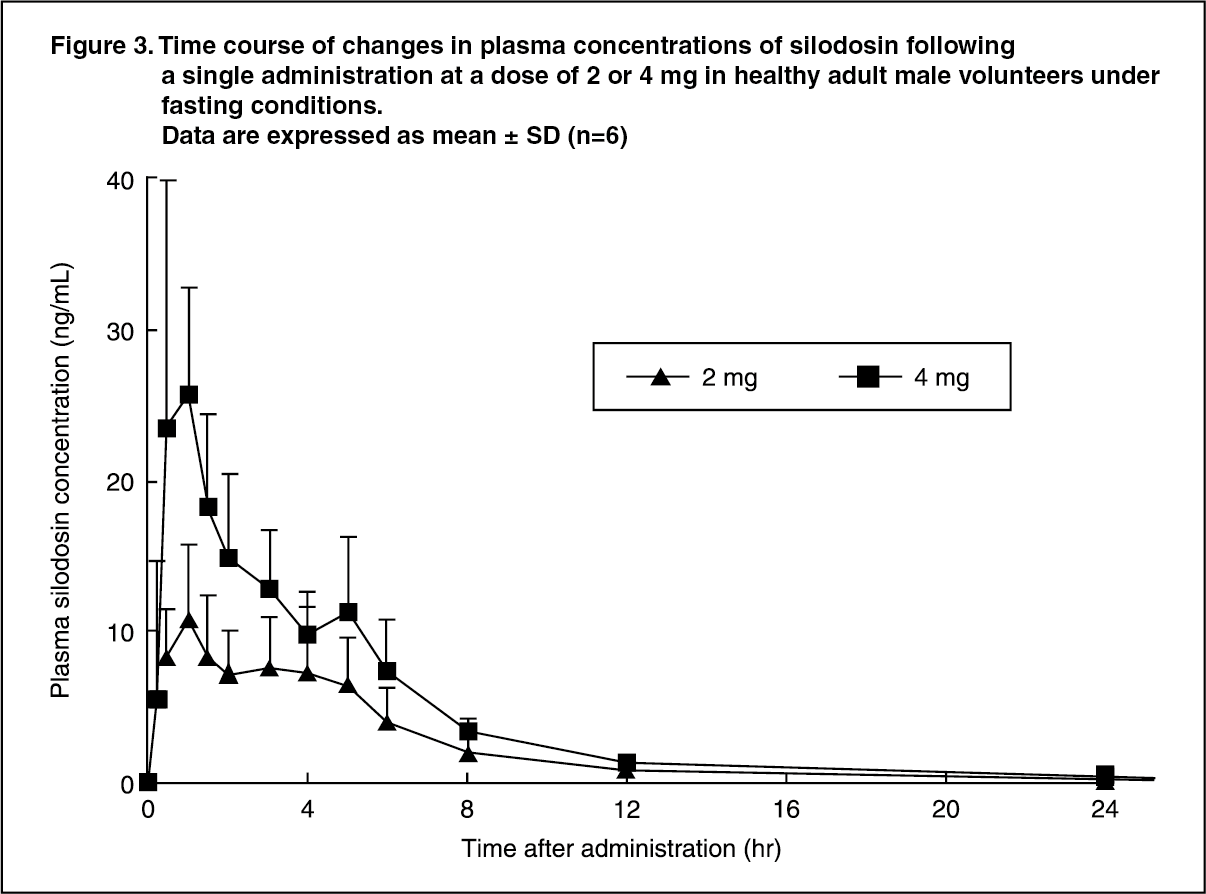
Click on icon to see table/diagram/imageSingle dose: When a single oral dose of this drug was administered to healthy adult male volunteers (6 subjects/group) at doses ranging from 0.5 to 12 mg, plasma concentrations of silodosin increased dose-dependently, and Cmax and AUC0-∞, showed linearity. The time course of changes in plasma concentrations of silodosin following a single oral administration of silodosin at a dose of 2 or 4 mg is shown in Figure 3. (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRepeated dose: When Silodosin was administered orally twice daily for 7 days (once daily on days 1 and 7) at a dose of 4 mg/dose in 5 healthy adult male volunteers, plasma concentrations of silodosin reached a steady state on day 3. The accumulation factor relative to the first dose was 1.1-fold. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetic parameters following repeated administration are shown as the results obtained from the time course of changes in concentration on day 7 less the cumulative concentration from days 0-6.
Elderly patients: When a single 4 mg dose of Silodosin was administered orally to 12 elderly males (age range: 65 to 75 year) postprandially, no obvious differences in the pharmacokinetic profile were observed compared to that in 9 non-elderly (age range: 21 to 31 years) males. The pharmacokinetic parameters in elderly males who received treatment with silodosin are shown in Table 6. (See Table 6.)
 Click on icon to see table/diagram/image
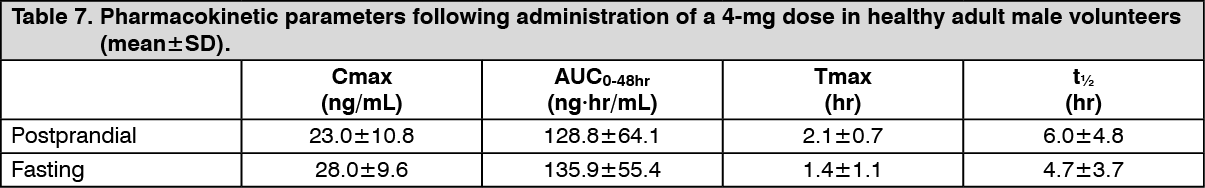
Click on icon to see table/diagram/imageWhen a single 4 mg dose of silodosin was administered orally to 11 healthy adult male volunteers 30 min postprandially or under fasting conditions, the Cmax, AUC0-48hr, Tmax, and t½ following postprandial administration (or under fasting conditions) were 23.0 (28.0) ng/mL, 128.8 (135.9) ng·hr/mL, 2.1 (1.4) hr, and 6.0 (4.7) hr, respectively (see Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe clearance and distribution volume following administration of silodosin solution (2 mg) to 11 healthy adult male volunteers by intravenous infusion over 4 hr were 167.0±33.8 mL/min and 49.5±17.3L, respectively.
The bioavailability following a single oral administration of silodosin at a dose of 4 mg was 32.2%.
Protein Binding: In an in vitro study, the human plasma-protein binding rate of silodosin was 95.6% (at a concentration of 100 ng/mL) and the main binding protein was α1-acid glycoprotein.
Metabolism and Excretion: Silodosin was metabolised mainly by CYP3A4, UGTs, ADH, and ALDH, with the major metabolites in plasma being a glucuronide and an oxidized metabolite of silodosin. When a single 8 mg dose of 14C-labeled silodosin solution was administered orally to 6 healthy male non-Japanese volunteers, the AUC0-12hr of silodosin and its glucuronide and oxidized metabolites relative to the AUC0-12hr of total radioactivity in plasma was 24.0, 21.9, and 34.9%, respectively. Other metabolites accounted for no more than 5%. In the 240-hour period after dosing 33.5 and 54.9% of administered radioactivity was excreted in urine and feces, respectively.
Elderly patients: The cumulative excretion in urine 0-48 hr after a single 4 mg dose of silodosin was administered orally to 12 elderly and 9 non-elderly male volunteers was 2.3 and 2.4% for silodosin, 1.6 and 1.8% for its glucuronide metabolite, and 4.5 and 4.9% for its oxidised metabolite, respectively.
Pharmacokinetics in Patients with Lower Urinary Tract Symptoms Associated with Benign Prostatic Hyperplasia: In an exploratory population pharmacokinetic analyses (n=258) of a long-term administration study with silodosin in patients with lower urinary tract symptoms associated with benign prostatic hyperplasia, the estimated plasma concentrations of silodosin (mean±SD) at steady state 2 and 12 hours post-dose were 24.8±8.0 and 7.4±3.3 ng/mL, respectively. An analysis of variable factors in relation to plasma concentrations of silodosin suggested that silodosin clearance is affected by body weight, age, CRP, ALT (GPT), and serum creatinine and distribution volume by body weight, age, CRP, and ALT (GPT). Of these factors, it was concluded that ALT (GPT) had the most effect on plasma concentrations of silodosin and it was suggested that, as a result of increased levels of ALT (GPT) (23→83 IU/L), silodosin clearance and distribution volume may decrease by approximately 47 and 27%, respectively.
Drug Interaction(s): Non-Japanese data: Ketoconazole (oral preparation not marketed in Japan) coadministration: When 16 healthy male volunteers (non-Japanese) who were receiving 200 mg of ketoconazole (p.o.) once daily for 4 days were coadministered a single 4 mg dose of silodosin (p.o.) on day 2, Cmax and AUC0-∞ of silodosin increased 3.7- and 2.9-fold, respectively, compared to when silodosin alone was administered.
Digoxin coadministration: When silodosin (4 mg, twice daily) was administered orally with digoxin (0.25 mg, once daily) for 8 days to 16 healthy male volunteers (non-Japanese), it was confirmed that silodosin has no effect on the pharmacokinetic profile of digoxin.
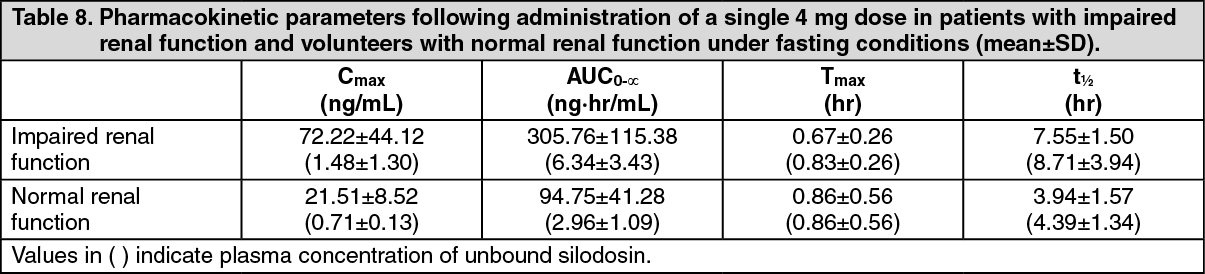
Pharmacokinetics in Patients with Impaired Renal Function: When a single 4 mg dose of silodosin was administered orally to 6 patients with impaired renal function (creatinine clearance: 27-49 mL/min) and 7 volunteers with normal renal function (creatinine clearance: 125-176 mL/min), the total plasma concentration of silodosin was increased (Cmax 3.1-fold; AUC0-∞: 3.2-fold) in patients with impaired renal function compared to that in the volunteer group. This increase in total plasma concentration of silodosin may be attributable to protein binding with serum α-1 acid glycoprotein, with a high correlation between total plasma concentration of silodosin and serum concentration of α1-acid glycoprotein observed. It should be noted that the increase in the plasma concentration of unbound silodosin (Cmax: 1.5-fold; AUC0-∞: 2.0-fold), which is considered to have a direct bearing on the manifestation of drug effect and incidence of adverse reactions associated with silodosin, was less than that for the total drug concentration (see Table 8).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image