



 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Evolocumab binds selectively to PCSK9 and prevents circulating PCSK9 from binding to the low density lipoprotein receptor (LDLR) on the liver cell surface, thus preventing PCSK9-mediated LDLR degradation. Increasing liver LDLR levels results in associated reductions in serum LDL-cholesterol (LDL-C).
Pharmacodynamic effects: In clinical trials, Evolocumab (Repatha) reduced unbound PCSK9, LDL-C, TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a), and increased HDL-C and ApoA1 in patients with primary hypercholesterolaemia and mixed dyslipidaemia.
A single subcutaneous administration of Evolocumab (Repatha) 140 mg or 420 mg resulted in maximum suppression of circulating unbound PCSK9 by 4 hours followed by a reduction in LDL-C reaching a mean nadir in response by 14 and 21 days, respectively. Changes in unbound PCSK9 and serum lipoproteins were reversible upon discontinuation of Evolocumab (Repatha). No increase in unbound PCSK9 or LDL-C above baseline was observed during the washout of evolocumab suggesting that compensatory mechanisms to increase production of PCSK9 and LDL-C do not occur during treatment.
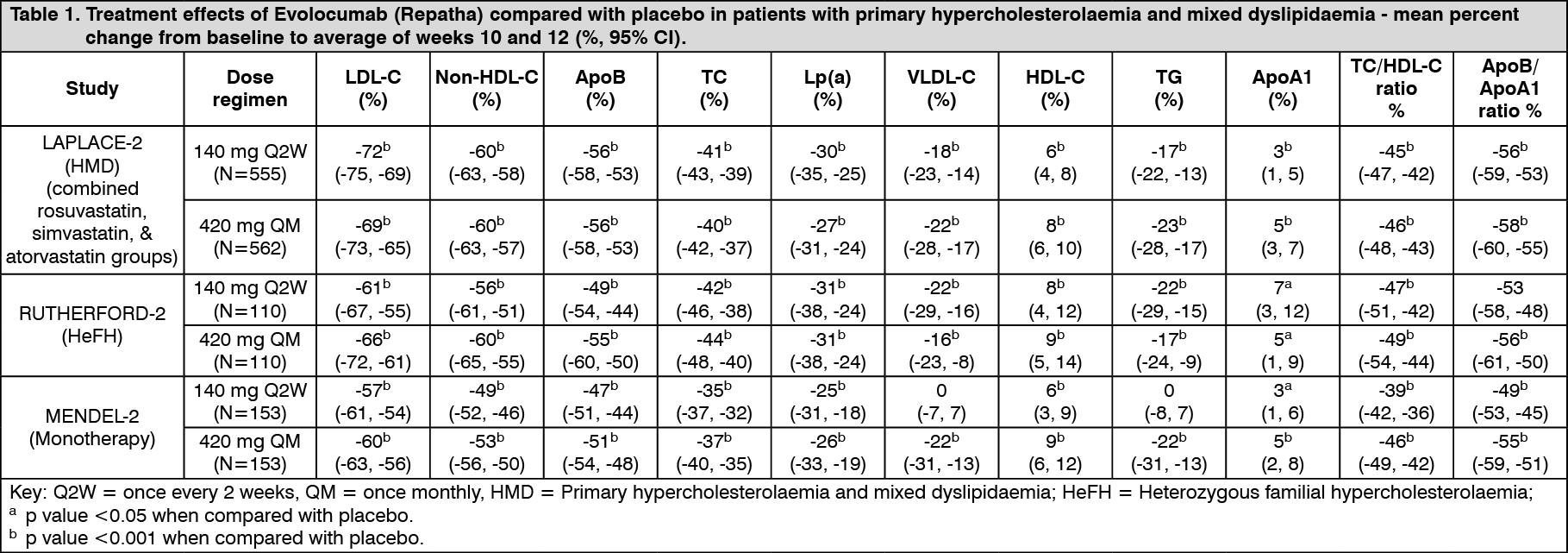
Subcutaneous regimens of 140 mg every 2 weeks and 420 mg once monthly were equivalent in average LDL-C lowering (mean of weeks 10 and 12) resulting in -72% to -57% from baseline compared with placebo. Treatment with Evolocumab (Repatha) resulted in a similar reduction of LDL-C when used alone or in combination with other lipid-lowering therapies.
Clinical efficacy in primary hypercholesterolaemia and mixed dyslipidaemia: LDL-C reduction of approximately 55% to 75% was achieved with Evolocumab (Repatha) as early as week 1 and maintained during long-term therapy. Maximal response was generally achieved within 1 to 2 weeks after dosing with 140 mg every 2 weeks and 420 mg once monthly. Evolocumab (Repatha) was effective in all subgroups relative to placebo and ezetimibe, with no notable differences observed between subgroups, such as age, race, gender, region, body-mass index, National Cholesterol Education Program risk, current smoking status, baseline coronary heart disease (CHD) risk factors, family history of premature CHD, glucose tolerance status, (i.e. diabetes mellitus type 2, metabolic syndrome, or neither), hypertension, statin dose and intensity, unbound baseline PCSK9, baseline LDL-C and baseline TG.
In 80-85% of all primary hyperlipidaemia patients treated with either dose, Evolocumab (Repatha) demonstrated a ≥ 50% reduction in LDL-C at the mean of weeks 10 and 12. Up to 99% of patients treated with either dose of Evolocumab (Repatha) achieved an LDL-C of < 2.6 mmol/L and up to 95% achieved an LDL-C < 1.8 mmol/L at the mean of weeks 10 and 12.
Combination with a statin and statin with other lipid-lowering therapies: LAPLACE-2 was an international, multicentre, double-blind, randomised, 12-week study in 1,896 patients with primary hypercholesterolaemia or mixed dyslipidaemia who were randomised to receive Evolocumab (Repatha) in combination with statins (rosuvastatin, simvastatin or atorvastatin).
Evolocumab (Repatha) was compared to placebo for the rosuvastatin and simvastatin groups and compared with placebo and ezetimibe for the atorvastatin group.
Evolocumab (Repatha) significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with placebo for the rosuvastatin and simvastatin groups and compared with placebo and ezetimibe for the atorvastatin group (p < 0.001). Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a) and increased HDL-C from baseline to mean of weeks 10 and 12 as compared to placebo for the rosuvastatin and simvastatin groups (p < 0.05) and significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), compared with placebo and ezetimibe for the atorvastatin group (p < 0.001) (see Tables 1 and 2).
RUTHERFORD-2 was an international, multicentre, double-blind, randomised, placebo-controlled, 12-week study in 329 patients with heterozygous familial hypercholesterolaemia on lipid-lowering therapies.
Evolocumab (Repatha) significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with placebo (p < 0.001). Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 VLDL-C, TG and Lp(a) and increased HDL-C and ApoA1 from baseline to mean of weeks 10 and 12 compared to placebo (p < 0.05) (see Table 1).
 Click on icon to see table/diagram/image
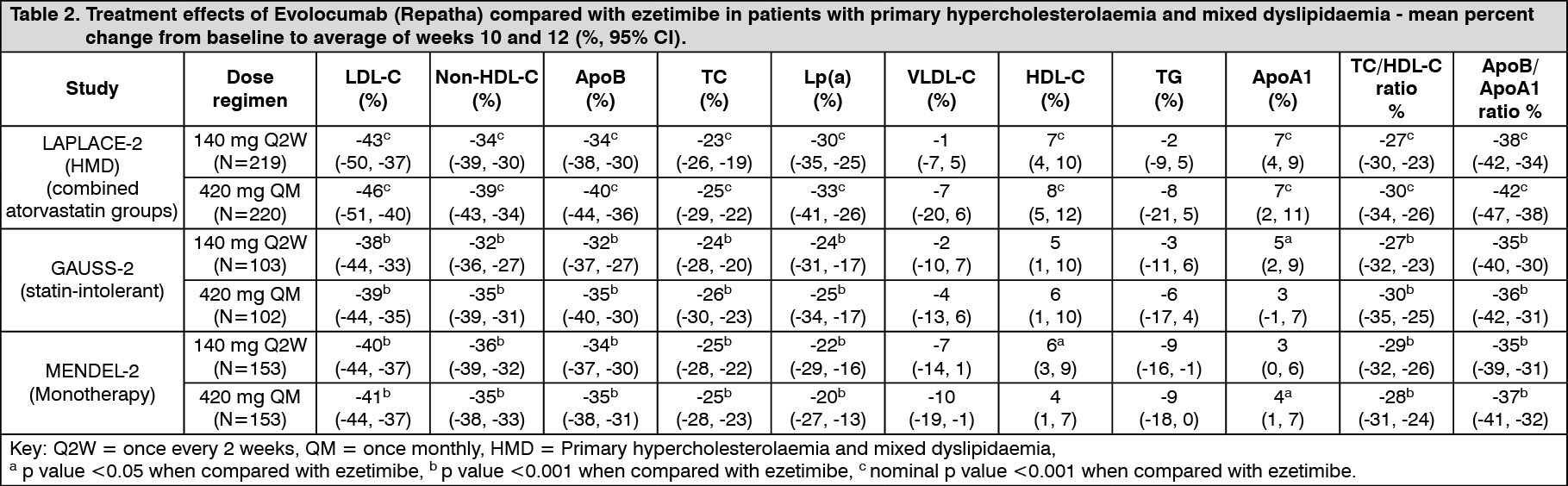
Click on icon to see table/diagram/imageStatin intolerant patients: GAUSS-2 was an international, multicentre, double-blind, randomised, ezetimibe-controlled, 12-week study in 307 patients who were statin-intolerant or unable to tolerate an effective dose of a statin. Evolocumab (Repatha) significantly reduced LDL-C compared with ezetimibe (p < 0.001). Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), from baseline to mean of weeks 10 and 12 compared to ezetimibe (p < 0.001) (see Table 2).
Monotherapy: MENDEL-2 was an international, multicentre, double-blind, randomised, placebo and ezetimibe-controlled, 12-week study of Evolocumab (Repatha) in 614 patients with primary hypercholesterolaemia and mixed dyslipidaemia. Evolocumab (Repatha) significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with both placebo and ezetimibe (p < 0.001). Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), from baseline to mean of weeks 10 and 12 compared with both placebo and ezetimibe (p < 0.001) (see Tables 1 and 2). (See Table 2.)
 Click on icon to see table/diagram/image
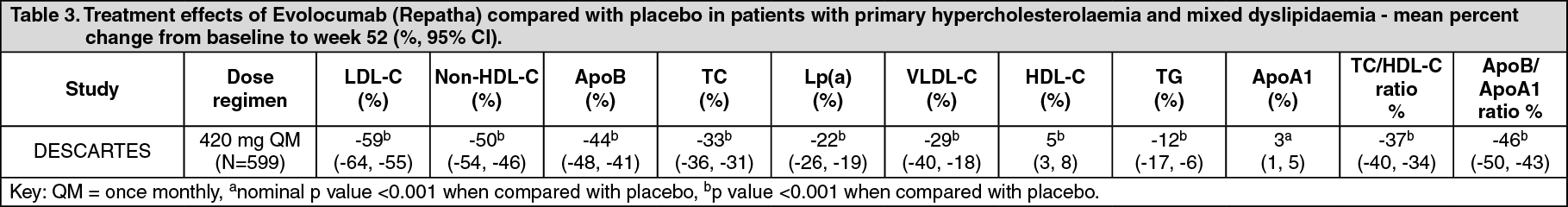
Click on icon to see table/diagram/imageLong-term efficacy in primary hypercholesterolaemia and mixed dyslipidaemia: DESCARTES was an international, multicentre, double-blind, randomised, placebo-controlled, 52-week study in 901 patients with hyperlipidaemia who received diet alone, atorvastatin, or a combination of atorvastatin and ezetimibe. Evolocumab (Repatha) 420 mg once monthly significantly reduced LDL-C from baseline at 52 weeks compared with placebo (p < 0.001). Treatment effects were sustained over 1 year as demonstrated by reduction in LDL-C from week 12 to week 52. Reduction in LDL-C from baseline at week 52 compared with placebo was consistent across background lipid-lowering therapies optimised for LDL-C and cardiovascular risk.
Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a), and increased HDL-C and ApoA1 at week 52 compared with placebo (p < 0.001). (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOSLER and OSLER-2 were two randomised, controlled, open-label extension studies to assess the long-term safety and efficacy of Evolocumab (Repatha) in patients who completed treatment in a 'parent' study. In each extension study, patients were randomised 2:1 to receive either Evolocumab (Repatha) plus standard of care (evolocumab group) or standard of care alone (control group) for the first year of the study. At the end of the first year (week 52 in OSLER and week 48 in OSLER-2), patients entered the all Evolocumab (Repatha) period in which all patients received open-label Evolocumab (Repatha) for either another 4 years (OSLER) or 2 years (OSLER-2).
A total of 1,324 patients enrolled in OSLER. Evolocumab (Repatha) 420 mg once monthly significantly reduced LDL-C from baseline at week 12 and week 52 compared with control (nominal p < 0.001). Treatment effects were maintained over 272 weeks as demonstrated by reduction in LDL-C from week 12 in the parent study to week 260 in the open-label extension. A total of 3,681 patients enrolled in OSLER-2. Evolocumab (Repatha) significantly reduced LDL-C from baseline at week 12 and week 48 compared with control (nominal p < 0.001). Treatment effects were maintained as demonstrated by reduction in LDL-C from week 12 to week 104 in the open-label extension. Evolocumab (Repatha) significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a), and increased HDL-C and ApoA1 from baseline to week 52 in OSLER and to week 48 in OSLER-2 compared with control (nominal p < 0.001). LDL-C and other lipid parameters returned to baseline within 12 weeks after discontinuation of Evolocumab (Repatha) at the beginning of OSLER or OSLER-2 without evidence of rebound.
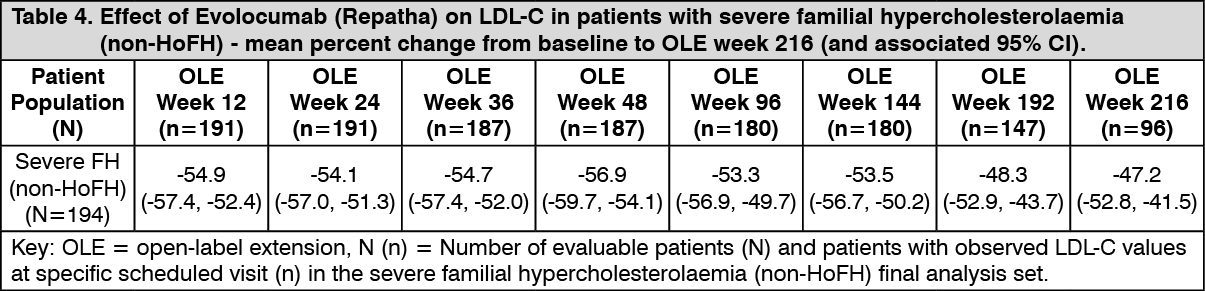
TAUSSIG was a multicentre, open-label, 5-year extension study to assess the long-term safety and efficacy of Evolocumab (Repatha), as an adjunct to other lipid lowering therapies, in patients with severe familial hypercholesterolaemia (FH), including homozygous familial hypercholesterolaemia. A total of 194 severe familial hypercholesterolaemia (non-HoFH) patients and 106 homozygous familial hypercholesterolaemia patients enrolled in TAUSSIG. All patients in the study were initially treated with Evolocumab (Repatha) 420 mg once monthly, except for those receiving lipid apheresis at enrolment who began with Evolocumab (Repatha) 420 mg once every 2 weeks. Dose frequency in non-apheresis patients could be titrated up to 420 mg once every 2 weeks based on LDL-C response and PCSK9 levels. Long-term use of Evolocumab (Repatha) demonstrated a sustained treatment effect as evidenced by reduction of LDL-C in patients with severe familial hypercholesterolaemia (non-HoFH) (see Table 4.)
Changes in other lipid parameters (TC, ApoB, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a sustained effect of long-term Evolocumab (Repatha) administration in patients with severe familial hypercholesterolaemia (non-HoFH). (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe long-term safety of sustained very low levels of LDL-C (i.e. < 0.65 mmol/L [< 25 mg/dL]) has not yet been established. Available data demonstrate that there are no clinically meaningful differences between the safety profiles of patients with LDL-C levels < 0.65 mmol/L and those with higher LDL-C, see Adverse Reactions.
Treatment of heterozygous familial hypercholesterolaemia in paediatric patients: HAUSER-RCT was a randomized, multicentre, placebo-controlled, double-blind, parallel-group, 24-week trial in 158 paediatric patients aged 10 to < 18 years with heterozygous familial hypercholesterolaemia. Patients were required to be on a low-fat diet and must have been receiving optimized background lipid-lowering therapy (statin at optimal dose, not requiring up titration). Enrolled patients were randomized in a 2:1 ratio to receive 24 weeks of subcutaneous once monthly 420 mg Evolocumab (Repatha) or placebo.
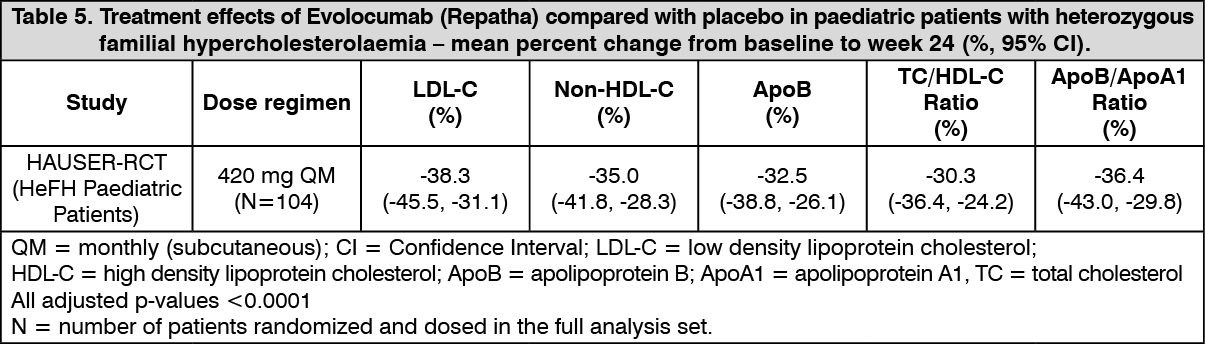
The primary efficacy endpoint in this trial was percent change from baseline to week 24 in LDL-C. The difference between Evolocumab (Repatha) and placebo in mean percent change in LDL-C from baseline to week 24 was 38% (95% CI: 45%, 31%; p < 0.0001). The least squares mean Standard Error (SE) reduction (p < 0.0001) in LDL-C from baseline at week 24 was 44% (2%) in the Evolocumab (Repatha) group and 6% (3%) in the placebo group. Mean absolute LDL-C values at week 24 were 104 mg/dL in the Evolocumab (Repatha) group and 172 mg/dL in the placebo group. Reductions in LDL-C were observed by the first post-baseline assessment at the week 12 time point and were maintained throughout the trial.
The secondary endpoint of this trial was mean percent change from baseline to weeks 22 and 24 in LDL-C, where week 22 reflects the peak and week 24 the trough of the subcutaneous once monthly dosing interval, and provides information about the time-averaged effect of Evolocumab (Repatha) therapy over the entire dosing interval. The least squares mean treatment difference between Evolocumab (Repatha) and placebo in mean percent change in LDL-C from baseline to the mean of week 22 and week 24 was 42% (95% CI: 48%, 36%; p < 0.0001). For additional results, see Table 5.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAUSER-OLE was an open-label, single-arm, multicentre, 80 week study of Evolocumab (Repatha) in 150 paediatric patients aged 10 to 17 years with HeFH that rolled-over from HAUSER-RCT and enrolled 13 de novo paediatric HoFH patients. Patients had to be on a low-fat diet and receiving background lipid-lowering therapy. All HeFH patients in this study received 420 mg Evolocumab (Repatha) subcutaneously once monthly (median exposure duration: 18.4 months). The mean (SE) percent changes in calculated LDL-C from baseline were: -44.4% (1.7%) at week 12, -41.0% (2.1%) at week 48, and -35.2% (2.5%) at week 80.
The mean (SE) percent change from baseline to week 80 in other lipid endpoints were: -32.1% (2.3%) non-HDL-C, -25.1% (2.3%) ApoB, -28.5% (2.0%) TC/HDL-C ratio, -30.3% (2.2%) ApoB/ApoA1 ratio, and -24.9% (1.9%) TC.
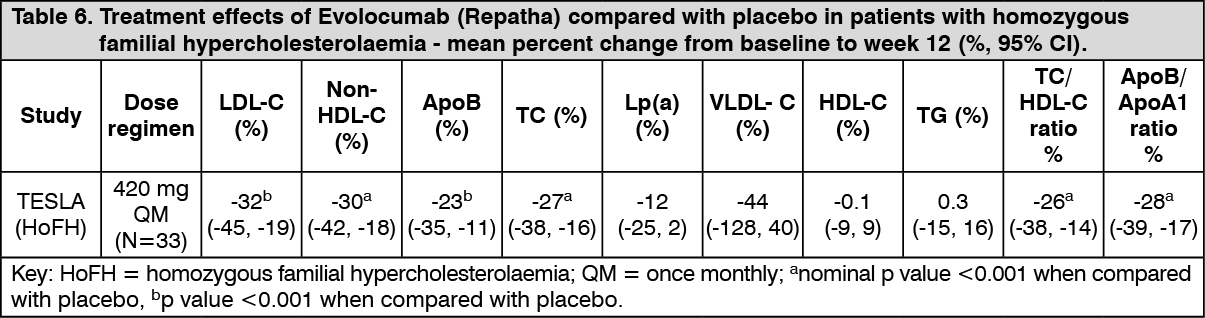
Treatment of homozygous familial hypercholesterolaemia: TESLA was an international, multicentre, double-blind, randomised, placebo-controlled 12-week study in 49 homozygous familial hypercholesterolaemia patients aged 12 to 65 years. Evolocumab (Repatha) 420 mg once monthly, as an adjunct to other lipid-lowering therapies (e.g., statins, bile-acid sequestrants), significantly reduced LDL-C and ApoB at week 12 compared with placebo (p < 0.001) (Table 6). Changes in other lipid parameters (TC, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a treatment effect of Evolocumab (Repatha) administration in patients with homozygous familial hypercholesterolaemia. (See Table 6.)
 Click on icon to see table/diagram/image
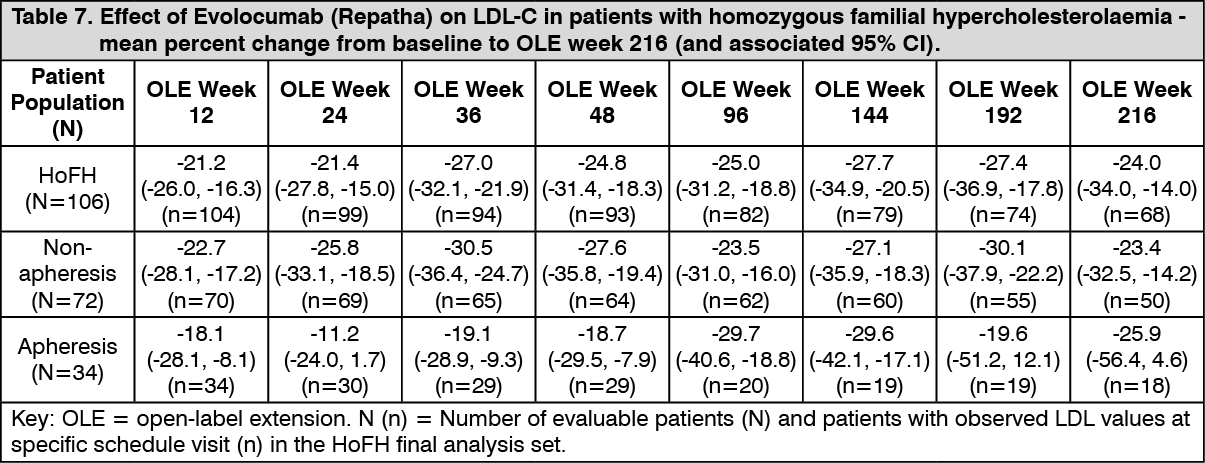
Click on icon to see table/diagram/imageLong-term efficacy in homozygous familial hypercholesterolaemia: In TAUSSIG, long-term use of Evolocumab (Repatha) demonstrated a sustained treatment effect as evidenced by reduction of LDL-C of approximately 20% to 30% in patients with homozygous familial hypercholesterolaemia not on apheresis and approximately 10% to 30% in patients with homozygous familial hypercholesterolaemia on apheresis (Table 7). Changes in other lipid parameters (TC, ApoB, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a sustained effect of long-term Evolocumab (Repatha) administration in patients with homozygous familial hypercholesterolaemia. Reductions in LDL-C and changes in other lipid parameters in 14 adolescent patients (aged ≥ 12 to < 18 years) with homozygous familial hypercholesterolaemia are comparable to those in the overall population of patients with homozygous familial hypercholesterolaemia. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAUSER-OLE was an open-label, single-arm, multicentre, 80-week trial in 12 HoFH subjects to evaluate the safety, tolerability and efficacy of Evolocumab (Repatha) for LDL-C reduction in paediatric patients from aged ≥ 10 to < 18 years of age with homozygous familial hypercholesterolaemia. Patients had to be on a low-fat diet and receiving background lipid-lowering therapy. All patients in the study received 420 mg Evolocumab (Repatha) subcutaneously once monthly. Median (Q1, Q3) LDL-C at baseline was 398 (343, 475) mg/dL. The median (Q1, Q3) percent change in LDL-C from baseline to week 80 was -14% (-41, 4). Reductions in LDL-C were observed by the first assessment at week 12 and was maintained throughout the trial, median (Q1, Q3) reductions ranging between 12% (-3, 32) and 15% (-4, 39). For additional results, please see Table 8.
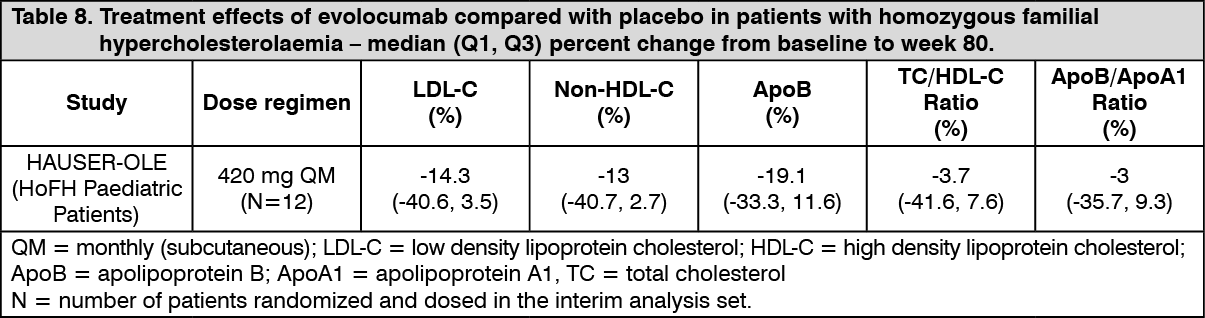 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffect on atherosclerotic disease burden: The effects of Evolocumab (Repatha) 420 mg once monthly on atherosclerotic disease burden, as measured by intravascular ultrasound (IVUS), were evaluated in a 78-week double-blind, randomised, placebo-controlled study in 968 patients with coronary artery disease on a stable background of optimal statin therapy. Evolocumab (Repatha) reduced both percent atheroma volume (PAV; 1.01% [95% CI 0.64, 1.38], p < 0.0001) and total atheroma volume (TAV; 4.89 mm3 [95% CI 2.53, 7.25], p < 0.0001) compared with placebo. Atherosclerotic regression was observed in 64.3% (95% CI 59.6, 68.7) and 47.3% (95% CI 42.6, 52.0) of patients who received Evolocumab (Repatha) or placebo respectively when measured by PAV. When measured by TAV, atherosclerotic regression was observed in 61.5% (95% CI 56.7, 66.0) and 48.9% (95% CI 44.2, 53.7) of patients who received Evolocumab (Repatha) or placebo respectively. The study did not investigate the correlation between atherosclerotic disease regression and cardiovascular events.
Effect on coronary atherosclerotic plaque morphology: The effects of Evolocumab (Repatha) 420 mg once monthly on coronary atherosclerotic plaques as assessed by optical coherence tomography (OCT), was evaluated in a 52-week double-blind, randomized, placebo-controlled study including adult patients initiated within 7 days of a non-ST-segment elevation acute coronary syndrome (NSTEACS) on maximally tolerated statin therapy. For the primary endpoint of absolute change in minimum FCT (fibrous cap thickness) in a matched segment of artery from baseline, least squares (LS) mean (95% CI) increased from baseline by 42.7 μm (32.4, 53.1) in the Evolocumab (Repatha) group and 21.5 μm (10.9, 32.1) in the placebo group, an additional 21.2 μm (4.7, 37.7) compared to placebo (p = 0.015; (38% difference (p = 0.041)). The reported secondary findings show treatment differences including change in mean minimum FCT (increase 32.5 μm (12.7, 52.4); p = 0.016) and absolute change in maximum lipid arc (-26° (-49.6, -2.4); p = 0.041).
Prevention of Cardiovascular Events: FOURIER was a phase 3, double-blind, randomised, placebo-controlled, event-driven, cardiovascular outcomes study to evaluate the effects of Evolocumab (Repatha) treatment in adult patients with established cardiovascular disease [prior myocardial infarction (81%), prior non-haemorrhagic stroke (19%), or symptomatic peripheral arterial disease (13%)].
Enrolled patients were on a stable background lipid lowering therapy and had LDL-C values ≥ 70 mg/dL (1.8 mmol/L) or non-HDL-C values ≥ 100 mg/dL (2.6 mmol/L) with at least one major or at least two minor risk factors. Most patients (99.7%) were on a high- (69.3%) or moderate-intensity (30.4%) statin therapy at baseline, and most patients (99.6%) were taking at least one other cardiovascular medication such as anti-platelet agents, beta blockers, ACE inhibitors, or angiotensin receptor blockers.
A total of 27,564 patients were randomised 1:1 to receive either Evolocumab (Repatha) (140 mg every 2 weeks or 420 mg once monthly) or placebo (every 2 weeks or once monthly, respectively) subcutaneously with regular assessments every 12 weeks. Patients were followed for a mean (SD) of 26.1 (6.4) months. A total of 24.6% of patients were female, 85.1% were white, 9.9% were Asian, 2.4% were Black, and 7.9% were Hispanic/Latino. The mean (SD) age was 62.5 (9.0) years. The median (Q1, Q3) LDL-C at baseline was 91.5 (79.5, 108.5) mg/dL (2.4 [2.0, 2.8] mmol/L).
Evolocumab (Repatha) significantly reduced the risk for the primary composite endpoint (time to cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina, or coronary revascularisation, whichever occurred first) and the key secondary composite endpoint (time to cardiovascular death, myocardial infarction, or stroke, whichever occurred first).
The results of primary and secondary efficacy endpoints are shown in Table 9 and Figures 1 and 2 as follows: See Table 9 and Figures 1 and 2.
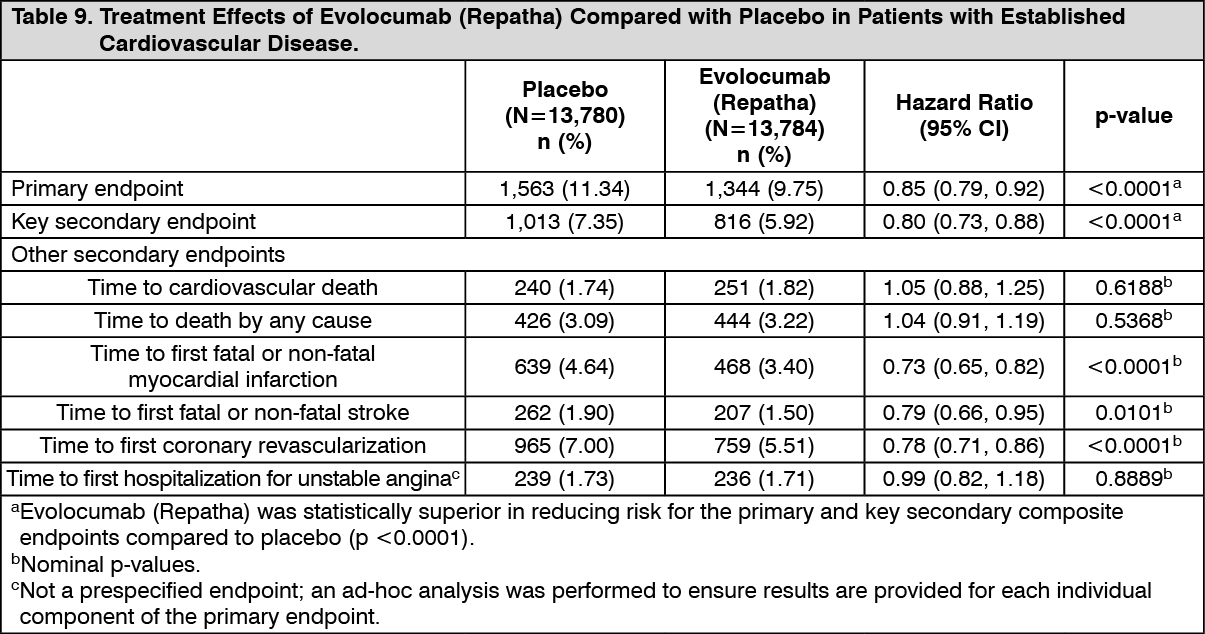 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image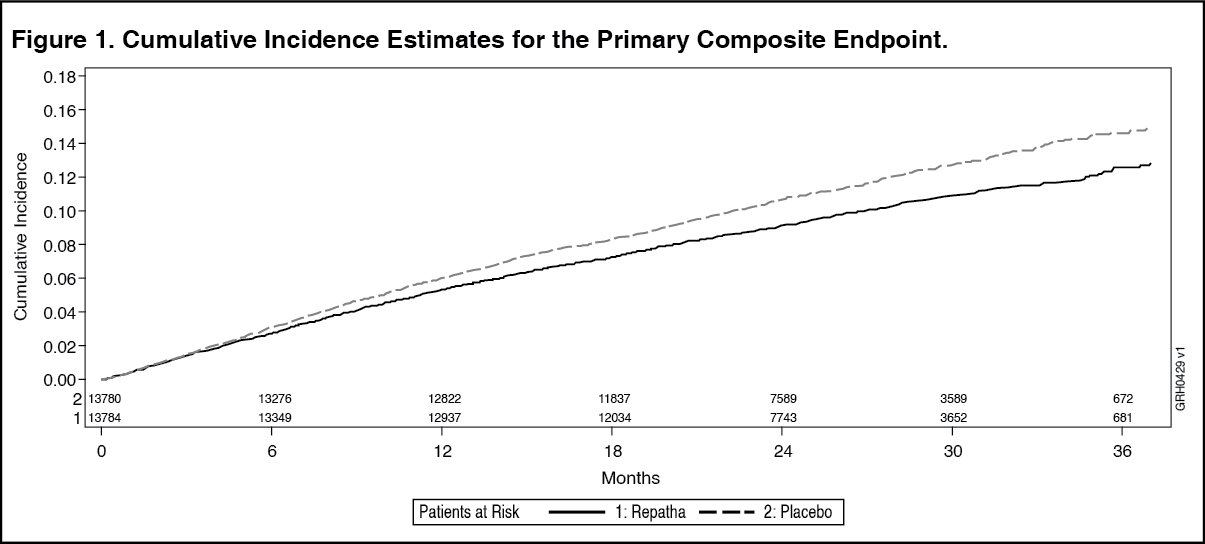 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image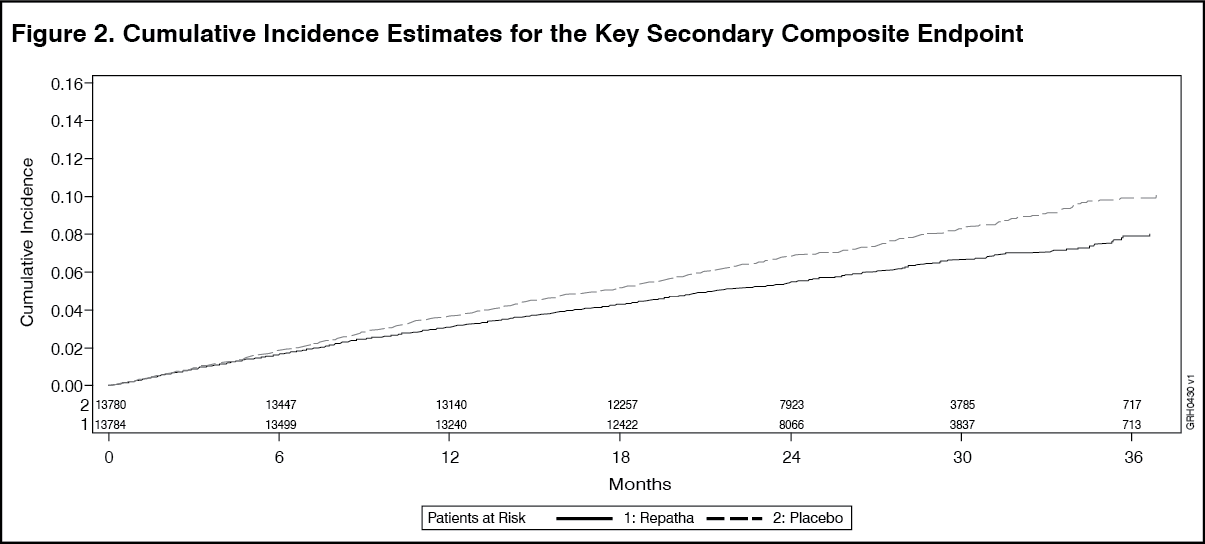 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe Kaplan-Meier curves for the primary and key secondary composite endpoints separated at approximately 5 months, and the magnitudes of the absolute risk reductions grew steadily over time.
In an exploratory landmark analysis of post-baseline subgroups, Evolocumab (Repatha) further reduced risk of the primary and key secondary composite endpoints more after the first year than in the first year of the study.
The efficacy of Evolocumab (Repatha) on the primary and key secondary composite endpoints was consistent across all pre-specified subgroups (e.g., baseline LDL-C, geographic region, age, sex, race, prior non-haemorrhagic stroke, symptomatic PAD, length of prior myocardial infarction, intensity of statin treatment at baseline, history of type 2 diabetes, ezetimibe use at baseline) relative to placebo.
Evolocumab (Repatha) reduced LDL-C by a median (Q1, Q3) of 63.8% (32.3%, 76.8%) to 69.5% (55.7%, 79.1%). The treatment difference in LDL-C reduction between Evolocumab (Repatha) and placebo ranged from 52.1% (95% CI: 49.2%, 55.0%) to 60.7% (95% CI: 60.1%, 61.3%). These reductions were maintained for more than 3 years. Corresponding median (Q1, Q3) LDL-C concentrations ranged from 29 (19, 43) mg/dL to 35 (21, 64) mg/dL in the Evolocumab (Repatha) group, and 25% of patients achieved a LDL-C concentration < 20 mg/dL.
Of the patients treated with Evolocumab (Repatha), 9,518 achieved at least one LDL-C value < 25 mg/dL. These patients had similar or lower incidence and similar type of adverse events, including neurocognitive events and new onset diabetes, compared to patients treated with Evolocumab (Repatha) or placebo who always had LDL-C ≥ 40 mg/dL. Lower LDL-C concentrations achieved during the study were associated with lower rates of cardiovascular events for the primary and secondary composite endpoint.
In a separate study of 1,974 patients with established cardiovascular disease enrolled in the FOURIER study, no clinically meaningful effect of Evolocumab (Repatha) was observed on cognitive function domains.
Other Supportive Clinical Information: In an integrated analysis of phase 2 and 3 randomised placebo and active controlled studies of Evolocumab (Repatha) for a duration of up to 52 weeks, adverse events were reported in 51% (N=1,609) of patients in the Evolocumab (Repatha) group who achieved an LDL < 25 mg/dL and 51% (N=2,565) of patients in the Evolocumab (Repatha) group who achieved an LDL < 40 mg/dL compared with 52% (N=1,339) of patients in the Evolocumab (Repatha) group with an LDL ≥ 40 mg/dL and 50% (N=2,038) of patients in the control group with LDL ≥ 40 mg/dL.
An integrated safety analysis of phase 2 and 3 randomised controlled studies of Evolocumab (Repatha) with statin therapy for a duration of up to 52 weeks was performed to assess alanine aminotransferase (ALT)/aspartate aminotransferase (AST) and creatine kinase (CK) for patients with normal values at baseline. The incidence of ALT or AST > 5x upper limit of normal was 0.1% in both the Evolocumab (Repatha) (N=2,523) and control (N=1,249) groups. In the same studies, CK > 10 x upper limit of normal was 0.2% (N=2,486) in the Evolocumab (Repatha) group and 0.1% (N=1,217) in the control group.
The safety of Evolocumab (Repatha) in the long-term, controlled studies was similar to the findings in the integrated analysis of phase 2 and 3 placebo and active controlled studies.
Effect on LDL-C during acute phase of Acute Coronary Syndromes (ACS): EVOPACS was a single country, multicentre, double-blind, randomized, placebo-controlled, 8-week study on 308 ACS patients with evolocumab initiated in-hospital within 24 to 72 hours of presentation.
If patients were not on a statin or were on statin treatment other than atorvastatin 40 mg prior to screening, this was stopped and atorvastatin 40 mg once daily was initiated. Randomisation was stratified by study centre and presence of stable statin treatment within ≥ 4 weeks prior to enrolment. Most subjects (241 [78%]) were not on stable statin treatment for ≥ 4 weeks prior to screening and most (235 [76%]) were not taking a statin at baseline. By week 4, 281 (97%) subjects were receiving high-intensity statins. Evolocumab (Repatha) 420 mg once monthly significantly reduced LDL-C from baseline to week 8 compared with placebo (p < 0.001). The mean (SD) reduction in calculated LDL-C from baseline at week 8 was 77.1% (15.8%) in the evolocumab group and 35.4% (26.6%) in the placebo group with a least squares (LS) mean difference (95% CI) of 40.7% (36.2%, 45.2%). Baseline LDL-C values were 3.61 mmol/L (139.5 mg/dL) in the evolocumab group and 3.42 mmol/L (132.2 mg/dL) in the placebo group. LDL-C reductions in this study were consistent with previous studies where Evolocumab (Repatha) was added to stable lipid-lowering therapy as demonstrated by on-treatment LDL-C levels at week 8 in this study (reflecting steady-state effect of high-intensity statin in both treatment arms) of 0.79 mmol/L (30.5 mg/dL) and 2.06 mmol/L (79.7 mg/dL) in the Evolocumab (Repatha) plus atorvastatin and the placebo plus atorvastatin groups, respectively.
The effects of Evolocumab (Repatha) in this patient population were consistent with those observed in previous studies in evolocumab clinical development program and no new safety concerns were noted.
Pharmacokinetics: Absorption and distribution: Following a single subcutaneous dose of 140 mg or 420 mg Evolocumab (Repatha) administered to healthy adults, median peak serum concentrations were attained in 3 to 4 days. Administration of single subcutaneous dose of 140 mg resulted in a Cmax mean (SD) of 13.0 (10.4) μg/mL and AUClast mean (SD) of 96.5 (78.7) day·μg/mL. Administration of single subcutaneous dose 420 mg resulted in a Cmax mean (SD) of 46.0 (17.2) μg/mL and AUClast mean (SD) of 842 (333) day·μg/mL. Three subcutaneous 140 mg doses were bioequivalent to a single subcutaneous 420 mg dose. The absolute bioavailability after SC dosing was determined to be 72% from pharmacokinetic models.
Following a single 420 mg Evolocumab (Repatha) intravenous dose, the mean (SD) steady-state volume of distribution was estimated to be 3.3 (0.5) L, suggesting evolocumab has limited tissue distribution.
Biotransformation: Evolocumab (Repatha) is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination: Evolocumab was estimated to have an effective half-life of 11 to 17 days.
In patients with primary hypercholesterolaemia or mixed dyslipidaemia on high dose statin, the systemic exposure of evolocumab was slightly lower than in subjects on low-to-moderate dose statin (the ratio of AUClast 0.74 [90% CI 0.29; 1.9]). An approximately 20% increase in the clearance is in part mediated by statins increasing the concentration of PCSK9 which did not adversely impact the pharmacodynamic effect of evolocumab on lipids. Population pharmacokinetic analysis indicated no appreciable differences in evolocumab serum concentrations in hypercholesterolaemic patients (non-familial hypercholesterolaemia or familial hypercholesterolaemia) taking concomitant statins.
Linearity/non-linearity: Following a single 420 mg intravenous dose, the mean (SD) systemic clearance was estimated to be 12 (2) mL/hr. In clinical studies with repeated subcutaneous dosing over 12 weeks, dose proportional increases in exposure were observed with dose regimens of 140 mg and greater. An approximate two to three-fold accumulation was observed in trough serum concentrations (Cmin (SD) 7.21 (6.6)) following 140 mg doses every 2 weeks or following 420 mg doses administered monthly (Cmin (SD) 11.2 (10.8)), and serum trough concentrations approached steady-state by 12 weeks of dosing.
No time dependent changes were observed in serum concentrations over a period of 124 weeks.
Renal impairment: No dose adjustment is necessary in patients with renal impairment. Data from the Evolocumab (Repatha) clinical trials did not reveal a difference in pharmacokinetics of evolocumab in patients with mild or moderate renal impairment relative to non-renally impaired patients.
In a clinical trial of 18 patients with either normal renal function (estimated glomerular filtration rate [eGFR] ≥ 90 mL/min/1.73 m2, n = 6), severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2, n = 6), or end-stage renal disease (ESRD) receiving haemodialysis (n = 6), exposure to unbound evolocumab as assessed by Cmax after a single 140 mg subcutaneous dose was decreased by 30% in patients with severe renal impairment and by 45% in patients with ESRD receiving haemodialysis. Exposure as assessed by AUClast was decreased by approximately 24% in patients with severe renal impairment and by approximately 45% in patients with ESRD receiving haemodialysis. The exact mechanism of PK differences is unknown; however, differences in body weight could not explain these differences. Some factors including small sample size and large inter-subject variability should be considered when interpreting the results. The pharmacodynamics and safety of evolocumab in patients with severe renal impairment and ESRD were similar to patients with normal renal function, and there were no clinically meaningful differences in LDL-C lowering. Therefore, no dose adjustments are necessary in patients with severe renal impairment or ESRD receiving haemodialysis.
Hepatic impairment: No dose adjustment is necessary in patients with mild hepatic impairment (Child-Pugh class A). Single 140 mg subcutaneous doses of Evolocumab (Repatha) were studied in 8 patients with mild hepatic impairment, 8 patients with moderate hepatic impairment and 8 healthy subjects. The exposure to evolocumab was found to be approximately 40-50% lower compared to healthy subjects. However, baseline PCSK9 levels and the degree and time course of PCSK9 neutralisation were found to be similar between patients with mild or moderate hepatic impairment and healthy volunteers. This resulted in similar time course and extent of absolute LDL-C lowering. Evolocumab (Repatha) has not been studied in patients with severe hepatic impairment (Child-Pugh class C) (see Precautions).
Body weight: Body weight was a significant covariate in population PK analysis impacting evolocumab trough concentrations, however there was no impact on LDL-C reduction. Following repeat subcutaneous administration of 140 mg every 2 weeks, the 12-week trough concentrations were 147% higher and 70% lower in patients of 69 kg and 93 kg respectively, than that of the typical 81 kg subject. Less impact from body weight was seen with repeated subcutaneous evolocumab 420 mg monthly doses.
Other special populations: Population pharmacokinetic analyses suggest that no dose adjustments are necessary for age, race or gender. The pharmacokinetics of evolocumab were influenced by body weight without having any notable effect on LDL-C lowering. Therefore, no dose adjustments are necessary based on body weight.
The pharmacokinetics of Evolocumab (Repatha) were evaluated in 103 paediatric patients aged ≥ 10 to < 18 years with heterozygous familial hypercholesterolaemia (HAUSER-RCT). Following subcutaneous administration of 420 mg Evolocumab (Repatha) once monthly, mean (SD) trough serum concentrations were 22.4 (14.7) mcg/mL, 64.9 (34.4) mcg/mL and 25.8 (19.2) mcg/mL over the Week 12, Week 22 and Week 24 time points, respectively. The pharmacokinetics of Evolocumab (Repatha) were evaluated in 12 paediatric patients aged ≥ 10 to < 18 years with homozygous familial hypercholesterolaemia (HAUSER-OLE). Following subcutaneous administration of 420 mg Evolocumab (Repatha) once monthly, mean (SD) serum trough concentrations were 20.3 (14.6) mcg/mL and 17.6 (28.6) mcg/mL at Week 12 and Week 80, respectively.
Toxicology: Preclinical safety data: Evolocumab was not carcinogenic in hamsters at exposures much higher than patients receiving evolocumab at 420 mg once monthly. The mutagenic potential of evolocumab has not been evaluated.
In hamsters and cynomolgus monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly, no effect on male or female fertility was observed.
In cynomolgus monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly, no effects on embryo-foetal or postnatal development (up to 6 months of age) were observed.
Apart from a reduced T-cell Dependent Antibody Response in cynomolgus monkeys immunised with keyhole limpet haemocyanin (KLH) after 3 months of treatment with evolocumab, no adverse effects were observed in hamsters (up to 3 months) and cynomolgus monkeys (up to 6 months) at exposures much higher than patients receiving evolocumab at 420 mg once monthly. The intended pharmacological effect of decreased serum LDL-C and total cholesterol were observed in these studies and was reversible upon cessation of treatment.
In combination with rosuvastatin for 3 months, no adverse effects were observed in cynomolgus monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly. Reductions in serum LDL-C and total cholesterol were more pronounced than observed previously with evolocumab alone, and were reversible upon cessation of treatment.