



 Sign Out
Sign Out
Pharmacology: Mechanism of Action: Sorafenib is a multikinase inhibitor that decreases tumor cell proliferation in vitro.
Sorafenib was shown to inhibit multiple intracellular (c-CRAF, BRAF and mutant BRAF) and cell surface kinases (KIT, FLT-3, RET, VEGFR-1, VEGFR-2, VEGFR-3 and PDGFR-β). Several of these kinases are thought to be involved in tumor cell signaling, angiogenesis and apoptosis. Sorafenib inhibited tumor growth of the human hepatocellular carcinoma and renal cell carcinoma, and several other human tumor xenografts in immunocompromised mice. A reduction in tumor angiogenesis and increases in tumor apoptosis was seen in models of human hepatocellular and renal cell carcinoma. Additionally a reduction in tumor cell signaling was seen in a model of human hepatocellular carcinoma.
Clinical Efficacy and Safety: The clinical safety and efficacy of Nexavar have been studied in patients with hepatocellular carcinoma (HCC) and in patients with advanced renal cell carcinoma (RCC).
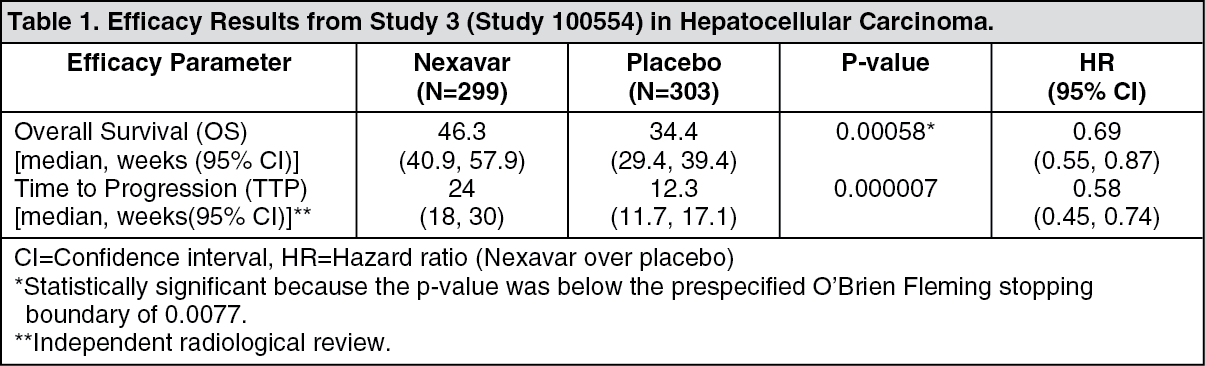
Hepatocellular Carcinoma: Study 3 (study 100554) was a phase III, international, multicenter, randomized, double-blind, placebo-controlled trial in 602 patients with hepatocellular carcinoma. Overall survival (OS) was a primary endpoint of this study, time to progression (TTP) a secondary endpoint.
Demographics and baseline disease characteristics were comparable between Nexavar and placebo groups with regard to age, gender, race, performance status, etiology (including hepatitis B, hepatitis C and alcoholic liver disease), TNM stage (stage I: <1% vs <1%; stage II: 10.4% vs 8.3%; stage III: 37.8% vs 43.6%; stage IV: 50.8% vs 46.9%), absence of both macroscopic vascular invasion and extrahepatic tumor spread (30.1% vs 30%), and BCLC stage (stage B: 18.1% vs 16.8%; stage C: 81.6% vs 83.2%; stage D: <1% vs 0%). Liver function Child-Pugh status was comparable between Nexavar and placebo groups (A: 95% vs 98%; B: 5% vs 2%). Only 1 patient with Child-Pugh C liver dysfunction was treated in the study. Prior treatment included surgical resection procedures (19.1% vs 20.5%), locoregional therapies (including radiofrequency ablation, percutaneous ethanol injection and transarterial chemoembolisation; 38.8% vs 40.6%), radiotherapy (4.3% vs. 5%) and systemic therapy (3% vs. 5%).
The study was stopped after a planned interim analysis of OS had crossed the prespecified efficacy boundary. This OS analysis showed a statistically significant advantage for Nexavar over placebo for OS (HR: 0.69, p=0.00058, see Table 1 and figure). This advantage was consistent across almost all subsets analyzed. In the prespecified stratification factors [Eastern Cooperative Oncology Group (ECOG) status, presence or absence of macroscopic vascular invasion and/or extrahepatic tumor spread and region] the hazard ratio consistently favored Nexavar over placebo. The time to tumor progression (TTP, by independent radiological review) was significantly larger in the Nexavar Arm (HR: 0.58, p=0.000007, see Table 1). (See Table 1 and figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image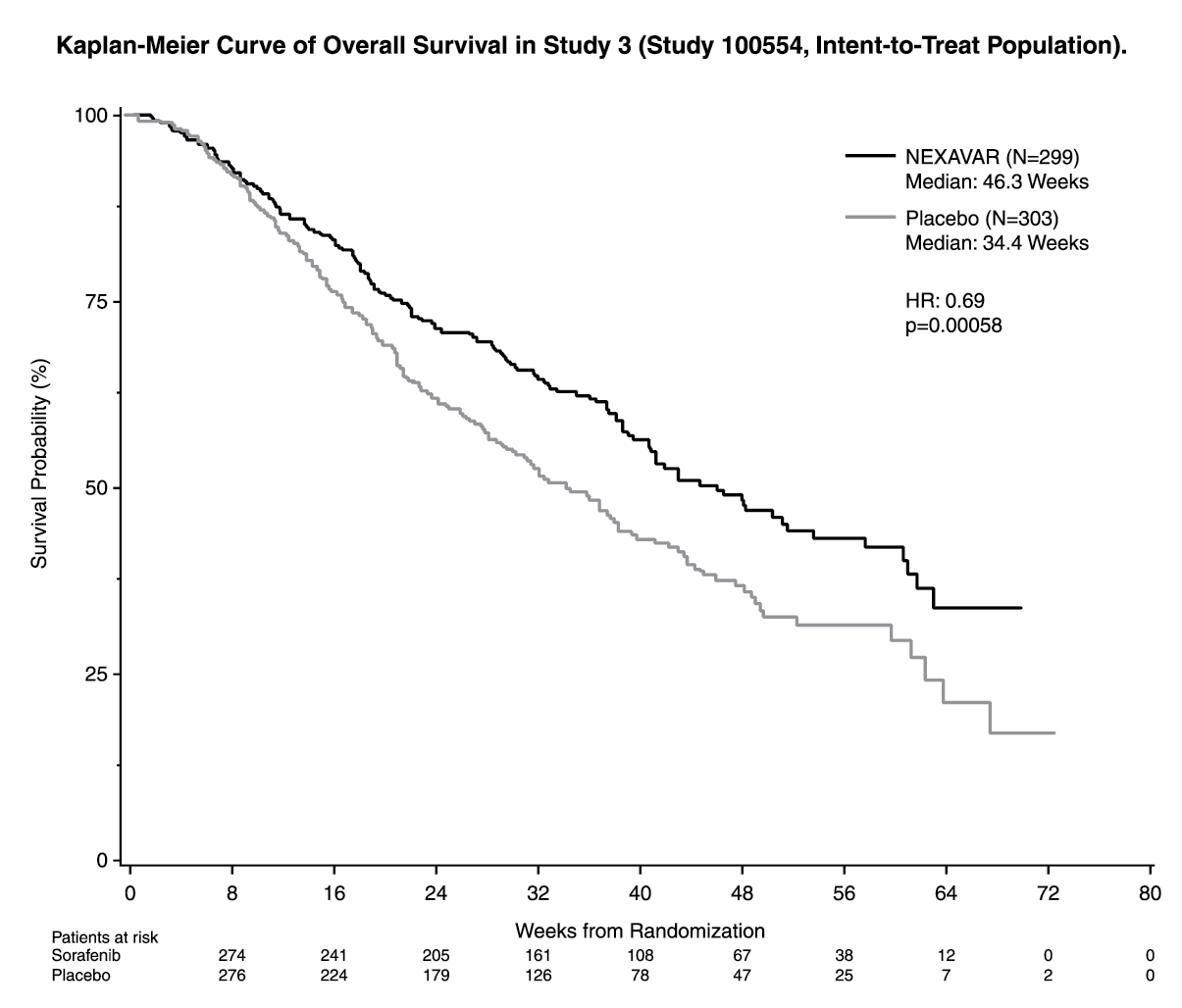 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRenal Cell Carcinoma: The safety and efficacy of Nexavar in the treatment of advanced RCC were studied in the following 2 randomized controlled clinical studies: Study 1 (11213) was a phase III, international, multicenter, randomized, double blind, placebo-controlled study in 903 patients. Primary study endpoints included overall survival and progression-free survival (PFS). Tumor response rate was a secondary endpoint.
Patients were randomized to Nexavar 400 mg twice daily (N=451) or to placebo (N=452). Baseline demographics and patient characteristics were well balanced for both treatment groups. Approximately half of the patients had an ECOG performance status of 0, and half of the patients were in the low Memorial Sloan Kettering Cancer Center (MSKCC) prognostic group.
In a planned interim analysis of survival based on 220 deaths, there was an estimated 39% improvement in overall survival for patients receiving sorafenib versus placebo. The estimated hazard ratio (risk of death with sorafenib compared to placebo) was 0.72 (95% CI, 0.55-0.95; p=0.018. The threshold for statistical significance of the interim analysis was p<0.0005).
The PFS analysis included 769 patients randomized to Nexavar 400 mg twice daily (N=384) or to placebo (N=385). PFS was evaluated by blinded independent radiological review using RECIST criteria. The median PFS was double for patients randomized to sorafenib (167 days) compared to placebo patients (84 days) (HR=0.44; 95% CI: 0.35-0.55; p<0.000001).
The effect on PFS was also explored across different patient subsets. The subsets included age above or below 65 years, ECOG PS 0 or 1, MSKCC prognostic risk category 1, whether the prior therapy was for progressive metastatic disease or for an earlier disease setting, and time from diagnosis of less than or >1.5 years. The effect of sorafenib on PFS was consistent across these subsets, including patients with no prior IL-2 or interferon therapy (n=137; 65 patients receiving sorafenib and 72 placebo), for whom the median PFS was 172 days on Sorafenib compared to 85 days on placebo.
Best overall tumor response was determined by investigator radiological review according to RECIST criteria. In the sorafenib group 1 patient (0.2%) had a complete response, 43 patients (9.5%) had a partial response, and 333 patients (73.8%) had stable disease. In the placebo group, 0 patients (0%) had complete response, 8 patients (1.8%) had partial response, and 239 patients (52.9%) had stable disease. Sorafenib demonstrated no overall deterioration in kidney-cancer specific symptoms (FKSI-10) or health-related quality of life compared to placebo. At 18 and 24 weeks of treatment, more patients receiving Sorafenib reported improvement in total FKSI-10 score (55 and 44%, respectively) and the physical well-being (FACT-G PWB) score (57 and 47%, respectively) versus placebo (FKSI-10, 33 and 21% and FACT-G PWB 37 and 21%, respectively).
Study 2 was a phase II randomized discontinuation trial in patients with metastatic malignancies, including RCC. The primary endpoint of the study was the percentage of randomized patients (N=65) remaining progression-free at 24 weeks. Progression-free survival was significantly longer in the sorafenib group (163 days) than in the placebo group (41 days) (p=0.0001, HR=0.29). The progression-free rate was significantly higher in patients randomized to sorafenib (50%) than in the placebo patients (18%) (p=0.0077).
QT Interval Prolongation: In a clinical pharmacology study, QT/QTc measurements were recorded in 31 patients at baseline (pre-treatment) and post-treatment. After one 28-day treatment cycle, at the time of maximum concentration of sorafenib, QTcB was prolonged by 4±19 msec and QTcF by 9±18 msec, as compared to placebo treatment at baseline. No subject showed a QTcB or QTcF >500 msec during the post-treatment ECG monitoring (see Precautions).
Pharmacokinetics: After administration of sorafenib tablets, the mean relative bioavailability is 38-49% when compared to an oral solution.
The elimination t½ of sorafenib is approximately 25-48 hrs. Multiple dosing of sorafenib for 7 days results in a 2.5- to 7-fold accumulation compared to single dose administration.
Steady-state plasma sorafenib concentrations are achieved within 7 days, with a peak to trough ratio of mean concentrations of <2.
Absorption and Distribution: Following oral administration, sorafenib reaches peak plasma levels in approximately 3 hrs. When given with a moderate-fat meal, bioavailability is similar to that in the fasted state. With a high-fat meal, sorafenib bioavailability is reduced by 29% compared to administration in the fasted state.
Mean Cmax and AUC increase less than proportionally beyond doses of 400 mg administered orally twice daily.
In vitro binding of sorafenib to human plasma proteins is 99.5%.
Metabolism and Elimination: Sorafenib is metabolized primarily in the liver undergoing oxidative metabolism, mediated by CYP3A4, as well as glucuronidation mediated by UGT1A9. Sorafenib conjugates may be cleaved in the gastrointestinal tract by bacterial glucuronidase activity, allowing reabsorption of unconjugated drug. Co-administration of neomycin interferes with this process, decreasing the mean bioavailability of sorafenib by 54%.
Sorafenib accounts for approximately 70-85% of the circulating analytes in plasma at steady state. Eight metabolites of sorafenib have been identified, of which 5 have been detected in plasma. The main circulating metabolite of sorafenib in plasma, the pyridine N-oxide, shows in vitro potency similar to that of sorafenib and comprises approximately 9-16% of circulating analytes at steady state.
Following oral administration of a 100 mg dose of a solution formulation of sorafenib, 96% of the dose was recovered within 14 days, with 77% of the dose excreted in feces and 19% of the dose excreted in urine as glucuronidated metabolites. Unchanged sorafenib, accounting for 51% of the dose, was found in feces but not in urine.
Studies on Enzyme Inhibition: Studies with human liver microsomes demonstrated that sorafenib is a competitive inhibitor of CYP2C19, CYP2D6 and CYP3A4. Concomitant clinical administration of midazolam, dextromethorphan, and omeprazole, which are substrates of cytochromes CYP3A4, CYP2D6, and CYP2C19, respectively, following 4 weeks of sorafenib administration did not alter the exposure of these agents. This indicates that sorafenib is neither an inhibitor nor an inducer of these cytochrome P-450 isoenzymes.
In vitro data show that sorafenib inhibits glucuronidation by the UGT1A1 (Ki=1 microM) and UGT1A9 (Ki=2 microM) pathways. Concomitant clinical administration of sorafenib with irinotecan, whose active metabolite SN-38 is further metabolized by the UGT1A1 pathway, resulted in a 67-120% increase in the AUC of SN-38. Systemic exposure to substrates of UGT1A1 and UGT1A9 may be increased when co-administered with sorafenib.
Sorafenib inhibits CYP2B6 and CYP2C8 in vitro with Ki values of 6 and 1-2 microM, respectively. Concomitant clinical administration of sorafenib with paclitaxel resulted in an increase, instead of a decrease, in the exposure of 6-OH paclitaxel, the active metabolite of paclitaxel that is formed by CYP2C8. These data suggest that sorafenib may not be an in vivo inhibitor of CYP2C8. Concomitant administration of sorafenib with cyclophosphamide resulted in a small decrease in cyclophosphamide exposure, but no decrease in the systemic exposure of 4-OH cyclophosphamide, the active metabolite of cyclophosphamide that is formed primarily by CYP2B6. These data suggest that sorafenib may not be an in vivo inhibitor of CYP2B6.
Studies with human liver microsomes demonstrated that sorafenib is a competitive inhibitor of CYP2C9 with a Ki value of 7-8 microM. The possible effect of sorafenib on a CYP2C9 substrate was assessed in patients receiving sorafenib or placebo in combination with warfarin. The mean changes from baseline in PT-INR were not higher in sorafenib patients compared to placebo patients, suggesting that sorafenib may not be an in vivo inhibitor of CYP2C9.
Special Populations: Elderly (>65 years) and Gender: Analyses of demographic data suggest that no dose adjustments are necessary for age or gender.
Pediatric Patients: There are no pharmacokinetic data in pediatric patients.
Hepatic Impairment: Sorafenib is cleared primarily by the liver.
In HCC patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment, exposure values were within the range observed in patients without hepatic impairment. The pharmacokinetics of sorafenib in Child-Pugh A and Child-Pugh B non-HCC patients were similar to the pharmacokinetics in healthy volunteers. The pharmacokinetics of sorafenib has not been studied in patients with severe (Child-Pugh C) hepatic impairment (see Dosage & Administration and Precautions).
Renal Impairment: In a clinical pharmacology study, the pharmacokinetics of sorafenib were evaluated following administration of a single 400 mg dose to subjects with normal renal function, and in subjects with mild (CrCl 50-80 mL/min), moderate (CrCl 30 to <50 mL/min), or severe (CrCl <30 mL/min) renal impairment, not requiring dialysis. There was no relationship observed between sorafenib exposure and renal function. No dosage adjustment is necessary based on mild, moderate or severe renal impairment not requiring dialysis (see Dosage & Administration).
Toxicology: Preclinical Safety Data: Systemic Toxicity: Repeat-dose toxicity studies revealed mild to moderate changes (degenerations and regenerations) in various organs.
After repeated dosing to young and growing dogs, effects on bone and teeth were observed. Changes consisted in irregular thickening of the femoral growth plate at a daily sorafenib dose of 600 mg/m2 body surface area (equivalent to 1.2 times the recommended clinical dose of 500 mg/m2 on a body surface area basis), hypocellularity of the bone marrow next to the altered growth plate at 200 mg/m2/day and alterations of the dentin composition at 600 mg/m2/day. Similar effects were not induced in adult dogs.
Positive genotoxic effects were obtained for sorafenib in an in vitro mammalian cell assay (Chinese hamster ovary) for clastogenicity (chromosome aberrations) in the presence of metabolic activation. One intermediate in the manufacturing process, which is also present in the final drug substance (<0.15%), was positive for mutagenesis in an in vitro bacterial cell assay (Ames test). Sorafenib was not genotoxic in the Ames test (the material contained the intermediate at 0.34%) and in an in vivo mouse micronucleus assay.
Genotoxicity and Carcinogenicity: Carcinogenicity studies have not been performed with sorafenib.
No specific studies with sorafenib have been conducted in animals to evaluate the effect on fertility. An adverse effect on male and female fertility can however be expected because repeat-dose studies in animals have shown changes in male and female reproductive organs. Typical changes consisted of signs of degeneration and retardation in testes, epididymides, prostate and seminal vesicles of rats, with clear effects at a daily sorafenib dose of 150 mg/m2 body surface area (equivalent to approximately 0.3 times the recommended clinical dose of 500 mg/m2 on a body surface area basis). Female rats showed central necrosis of the corpora lutea and arrested follicular development in the ovaries with a lowest observed effect at 30 mg/m2/day. Dogs showed tubular degeneration in the testes at 600 mg/m2/day and oligospermia at 1200 mg/m2/day.
Reproduction Toxicity: Sorafenib has been shown to be embryotoxic and teratogenic when administered to rats and rabbits. Observed effects included decreases in maternal and fetal body weights, an increased number of fetal resorptions and an increased number of external and visceral malformations. Adverse fetal outcomes were observed at an oral dose of 6 mg/m2/day in rats and 36 mg/m2/day in rabbits (see Use in pregnancy and Use in lactation under Precautions).