



 Sign Out
Sign Out
The cholesterol content of the liver is derived predominantly from three sources. The liver can synthesize cholesterol, take up cholesterol from the blood from circulating lipoproteins, or take up cholesterol absorbed by the small intestine.
Intestinal cholesterol is derived primarily from cholesterol secreted in the bile and from dietary cholesterol.
Ezetimibe has a mechanism of action that differs from those of other classes of cholesterol-reducing compounds (statins, bile acid sequestrants [resins], fibric acid derivatives, and plant stanols). The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols.
Ezetimibe does not inhibit cholesterol synthesis in the liver, or increase bile acid excretion. Instead, ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in clearance of cholesterol from the blood; this distinct mechanism is complementary to that of statins and of fenofibrate [see Clinical Studies as follows].
Pharmacodynamics: Clinical studies have demonstrated that elevated levels of total-C, LDL-C and Apo B, the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of HDL-C are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis. The independent effect of raising HDL-C or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined.
Ezetimibe reduces total-C, LDL-C, Apo B, non-HDL-C, and TG, and increases HDL-C in patients with hyperlipidemia. Administration of ezetimibe with a statin is effective in improving serum total-C, LDL-C, Apo B, non-HDL-C, TG, and HDL-C beyond either treatment alone. Administration of ezetimibe with fenofibrate is effective in improving serum total-C, LDL-C, Apo B, and non-HDL-C in patients with mixed hyperlipidemia as compared to either treatment alone. The effects of ezetimibe given either alone or in addition to a statin or fenofibrate on cardiovascular morbidity and mortality have not been established.
Clinical Studies: Primary Hyperlipidemia: Ezetimibe reduces total-C, LDL-C, Apo B, non-HDL-C, and TG, and increases HDL-C in patients with hyperlipidemia. Maximal to near maximal response is generally achieved within 2 weeks and maintained during chronic therapy.
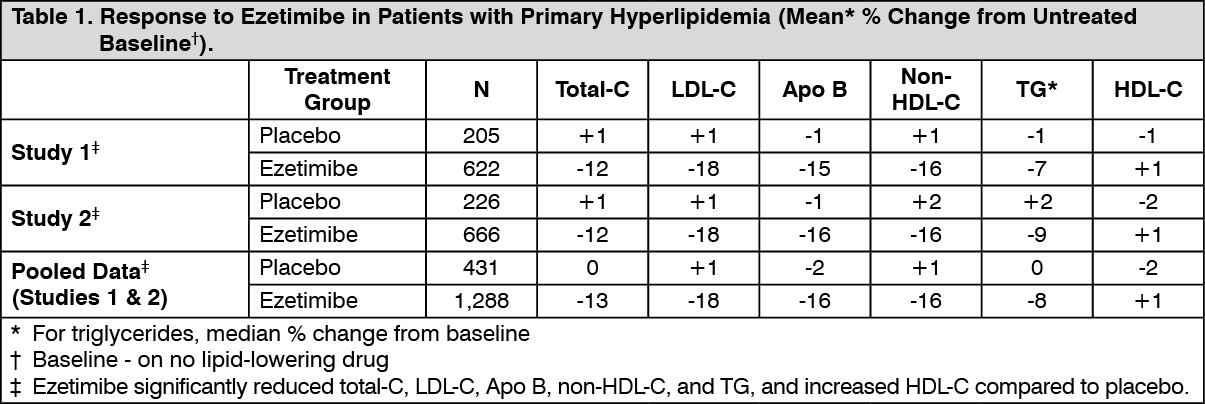
Monotherapy: In two multicenter, double-blind, placebo-controlled, 12-week studies in 1,719 patients with primary hyperlipidemia, ezetimibe significantly lowered total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared to placebo (see Table 1). Reduction in LDL-C was consistent across age, sex, and baseline LDL-C. (See Table 1.)
 Click on icon to see table/diagram/image
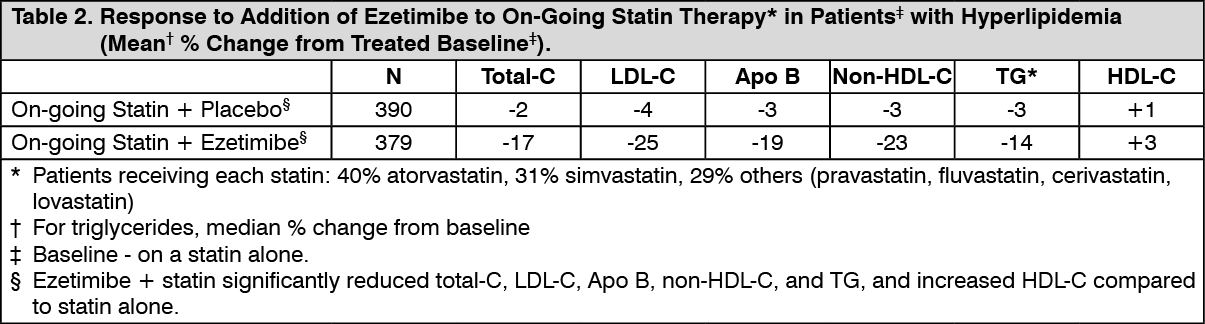
Click on icon to see table/diagram/imageCombination with Statins: Ezetimibe Added to On-going Statin Therapy: In a multicenter, double-blind, placebo-controlled, 8-week study, 769 patients with primary hyperlipidemia, known coronary heart disease or multiple cardiovascular risk factors who were already receiving statin monotherapy, but who had not met their NCEP ATP II target LDL-C goal were randomized to receive either ezetimibe or placebo in addition to their on-going statin.
Ezetimibe, added to on-going statin therapy, significantly lowered total-C, LDL-C, Apo B, non-HDL-C, and TG, and increased HDL-C compared with a statin administered alone (see Table 2). LDL-C reductions induced by ezetimibe were generally consistent across all statins. (See Table 2.)
 Click on icon to see table/diagram/image
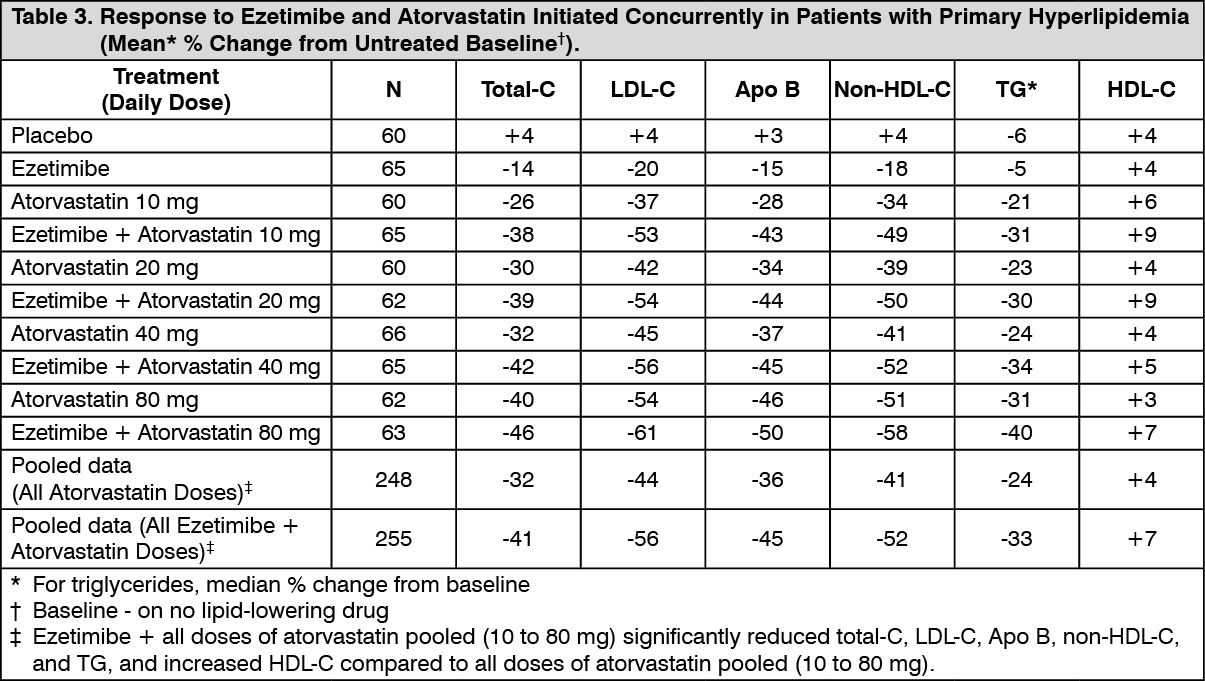
Click on icon to see table/diagram/imageEzetimibe Initiated Concurrently with a Statin: In four multicenter, double-blind, placebo-controlled, 12-week trials, in 2,382 hyperlipidemic patients, ezetimibe or placebo was administered alone or with various doses of atorvastatin, simvastatin, pravastatin, or lovastatin.
When all patients receiving ezetimibe with a statin were compared to all those receiving the corresponding statin alone, ezetimibe significantly lowered total-C, LDLC, Apo B, non-HDL-C, and TG, and, with the exception of pravastatin, increased HDL-C compared to the statin administered alone. LDL-C reductions induced by ezetimibe were generally consistent across all statins. (See footnote ‡, Tables 3 to 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image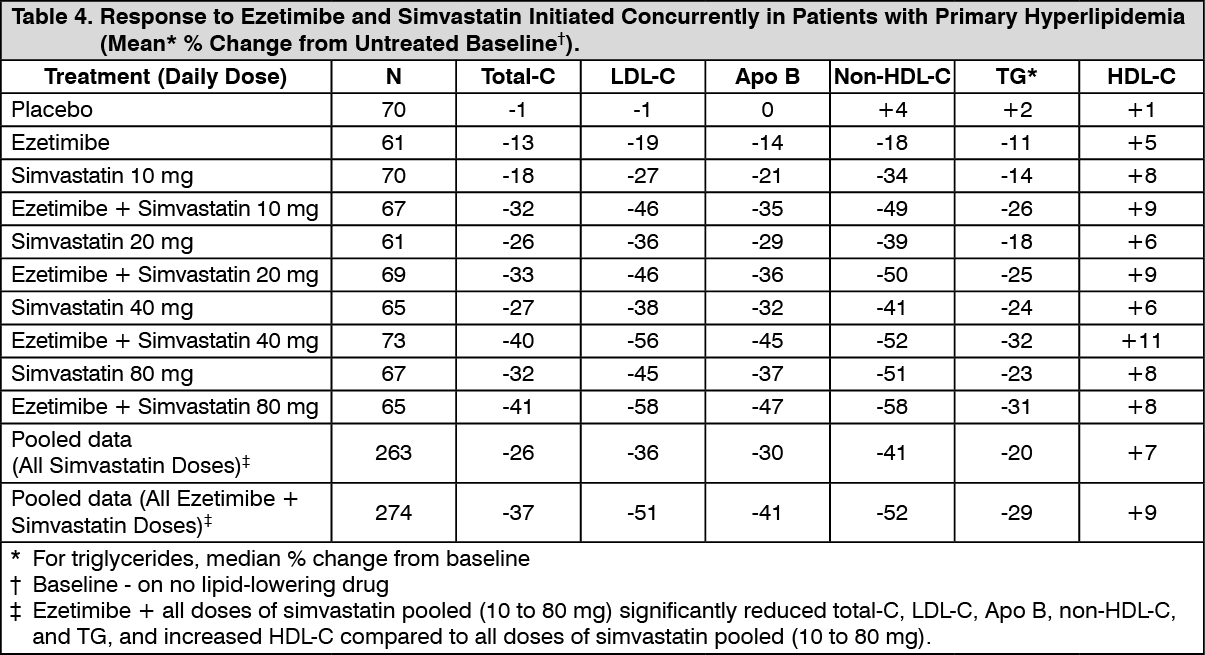 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image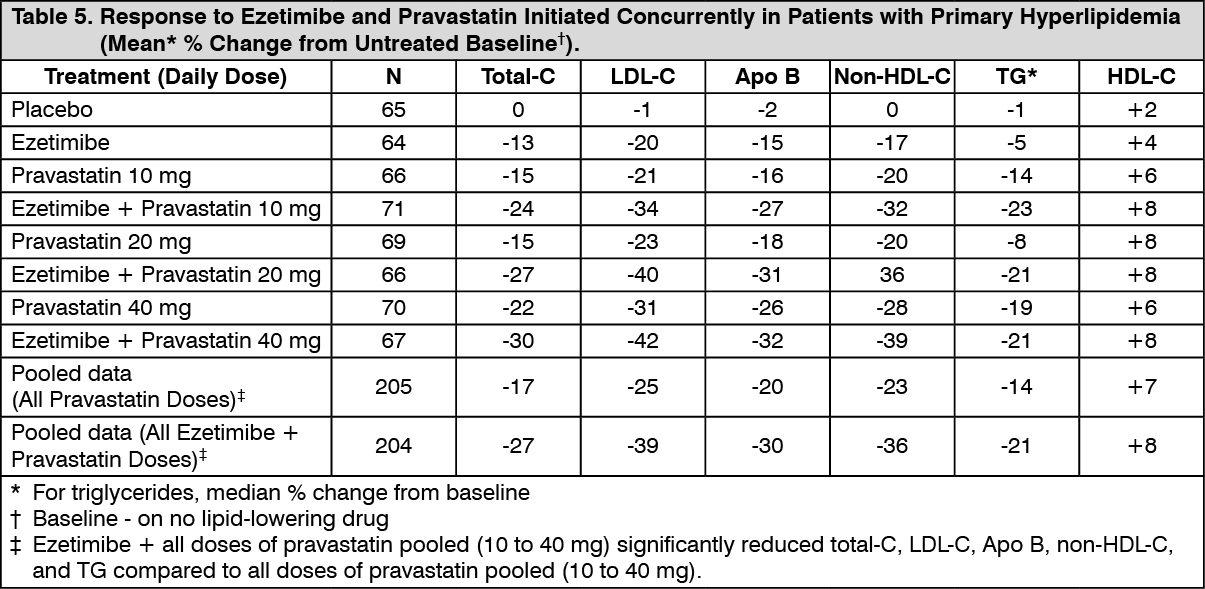 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image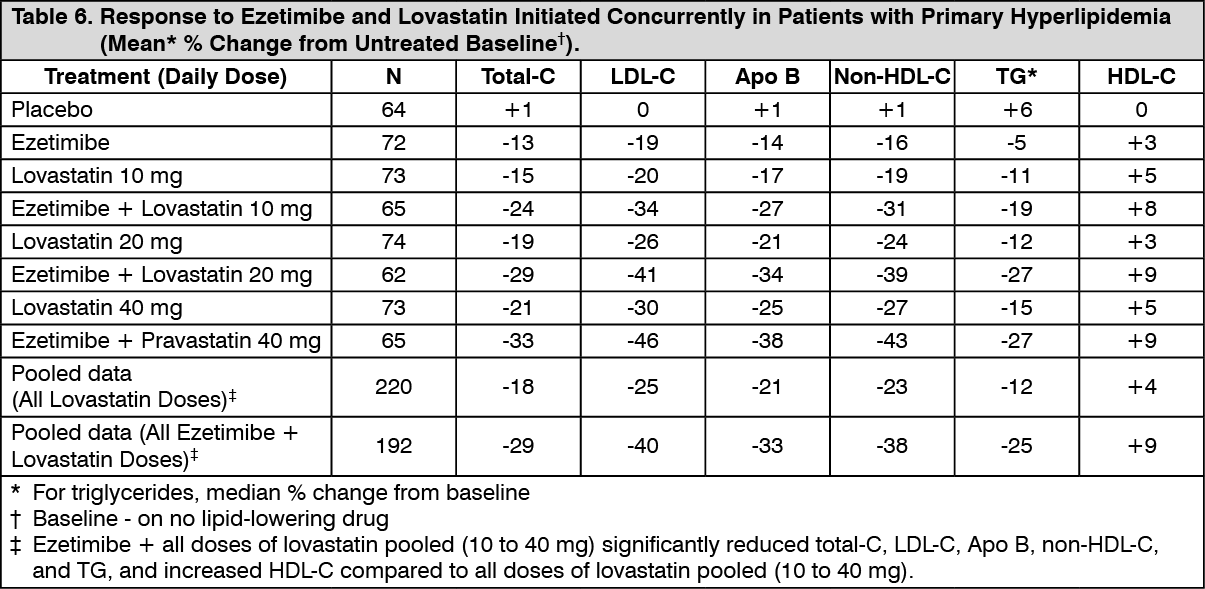 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCombination with Fenofibrate: In a multicenter, double-blind, placebo-controlled, clinical study in patients with mixed hyperlipidemia, 625 patients were treated for up to 12 weeks and 576 for up to an additional 48 weeks. Patients were randomized to receive placebo, ezetimibe alone, 160 mg fenofibrate alone, or ezetimibe and 160 mg fenofibrate in the 12-week study. After completing the 12-week study, eligible patients were assigned to ezetimibe co-administered with fenofibrate or fenofibrate monotherapy for an additional 48 weeks.
Ezetimibe co-administered with fenofibrate significantly lowered total-C, LDL-C, Apo B, and non-HDL-C compared to fenofibrate administered alone. The percent decrease in TG and percent increase in HDL-C for ezetimibe co-administered with fenofibrate were comparable to those for fenofibrate administered alone (see Table 7).
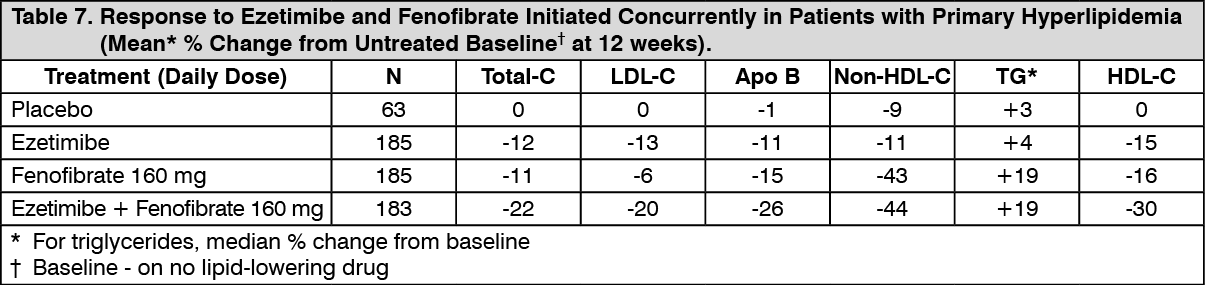 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe changes in lipid endpoints after an additional 48 weeks of treatment with ezetimibe co-administered with fenofibrate or with fenofibrate alone were consistent with the 12-week data displayed previously.
Homozygous Familial Hypercholesterolemia (HoFH): A study was conducted to assess the efficacy of ezetimibe in the treatment of HoFH. This double-blind, randomized, 12-week study enrolled 50 patients with a clinical and/or genotypic diagnosis of HoFH, with or without concomitant LDL apheresis, already receiving atorvastatin or simvastatin (40 mg). Patients were randomized to one of three treatment groups, atorvastatin or simvastatin (80 mg), ezetimibe administered with atorvastatin or simvastatin (40 mg), or ezetimibe administered with atorvastatin or simvastatin (80 mg). Due to decreased bioavailability of ezetimibe in patients concomitantly receiving cholestyramine [see Interactions], ezetimibe was dosed at least 4 hours before or after administration of resins. Mean baseline LDL-C was 341 mg/dL in those patients randomized to atorvastatin 80 mg or simvastatin 80 mg alone and 316 mg/dL in the group randomized to ezetimibe plus atorvastatin 40 or 80 mg or simvastatin 40 or 80 mg. Ezetimibe, administered with atorvastatin or simvastatin (40 and 80 mg statin groups, pooled), significantly reduced LDL-C (21%) compared with increasing the dose of simvastatin or atorvastatin monotherapy from 40 to 80 mg (7%). In those treated with ezetimibe plus 80 mg atorvastatin or with ezetimibe plus 80 mg simvastatin, LDL-C was reduced by 27%.
Limitations of Use: The effect of ezetimibe on cardiovascular morbidity and mortality has not been determined.
Pharmacokinetics: Absorption: After oral administration, ezetimibe is absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide). After a single 10 mg dose of ezetimibe to fasted adults, mean ezetimibe peak plasma concentrations (Cmax) of 3.4 to 5.5 ng/mL were attained within 4 to 12 hours (Tmax). Ezetimibe-glucuronide mean C values of 45 to 71 ng/mL were achieved between 1 and 2 hours (Tmax). There was no substantial deviation from dose proportionality between 5 and 20 mg. The absolute bioavailability of ezetimibe cannot be determined, as the compound is virtually insoluble in aqueous media suitable for injection.
Effect of Food on Oral Absorption: Concomitant food administration (high-fat or non-fat meals) had no effect on the extent of absorption of ezetimibe when administered as ezetimibe 10 mg tablets. The C value of ezetimibe was increased by 38% with consumption of max high-fat meals. Ezetimibe can be administered with or without food.
Distribution: Ezetimibe and ezetimibe-glucuronide are highly bound (>90%) to human plasma proteins.
Metabolism and Excretion: Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary and renal excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated.
In humans, ezetimibe is rapidly metabolized to ezetimibe-glucuronide. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are eliminated from plasma with a half-life of approximately 22 hours for both ezetimibe and ezetimibe-glucuronide. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling.
Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe (ezetimibe + ezetimibe-glucuronide) accounted for approximately 93% of the total radioactivity in plasma. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. Ezetimibe was the major component in feces and accounted for 69% of the administered dose, while ezetimibe-glucuronide was the major component in urine and accounted for 9% of the administered dose.
Special Populations: Geriatric Patients: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were about 2-fold higher in older (≥65 years) healthy subjects compared to younger subjects.
Pediatric Patients: [See Use in Children under Precautions].
Gender: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20%) in women than in men.
Race: Based on a meta-analysis of multiple-dose pharmacokinetic studies, there were no pharmacokinetic differences between Black and Caucasian subjects. Studies in Asian subjects indicated that the pharmacokinetics of ezetimibe were similar to those seen in Caucasian subjects.
Hepatic Impairment: After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh score 5 to 6), compared to healthy subjects. The mean AUC values for total ezetimibe and ezetimibe were increased approximately 3- to 4-fold and 5- to 6-fold, respectively, in patients with moderate (Child-Pugh score 7 to 9) or severe hepatic impairment (Child-Pugh score 10 to 15). In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment, the mean AUC values for total ezetimibe and ezetimibe were increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe hepatic impairment, ezetimibe is not recommended in these patients [see Precautions].
Renal Impairment: After a single 10 mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 mL/min/1.73 m2), the mean AUC values for total ezetimibe, ezetimibe-glucuronide, and ezetimibe were increased approximately 1.5-fold, compared to healthy subjects (n=9).
Drug Interactions [See Interactions]: Ezetimibe had no significant effect on a series of probe drugs (caffeine, dextromethorphan, tolbutamide, and IV midazolam) known to be metabolized by cytochrome P450 (1A2, 2D6, 2C8/9 and 3A4) in a "cocktail" study of twelve healthy adult males. This indicates that ezetimibe is neither an inhibitor nor an inducer of these cytochrome P450 isozymes, and it is unlikely that ezetimibe will affect the metabolism of drugs that re metabolized by these enzymes. (See Tables 8 and 9.)
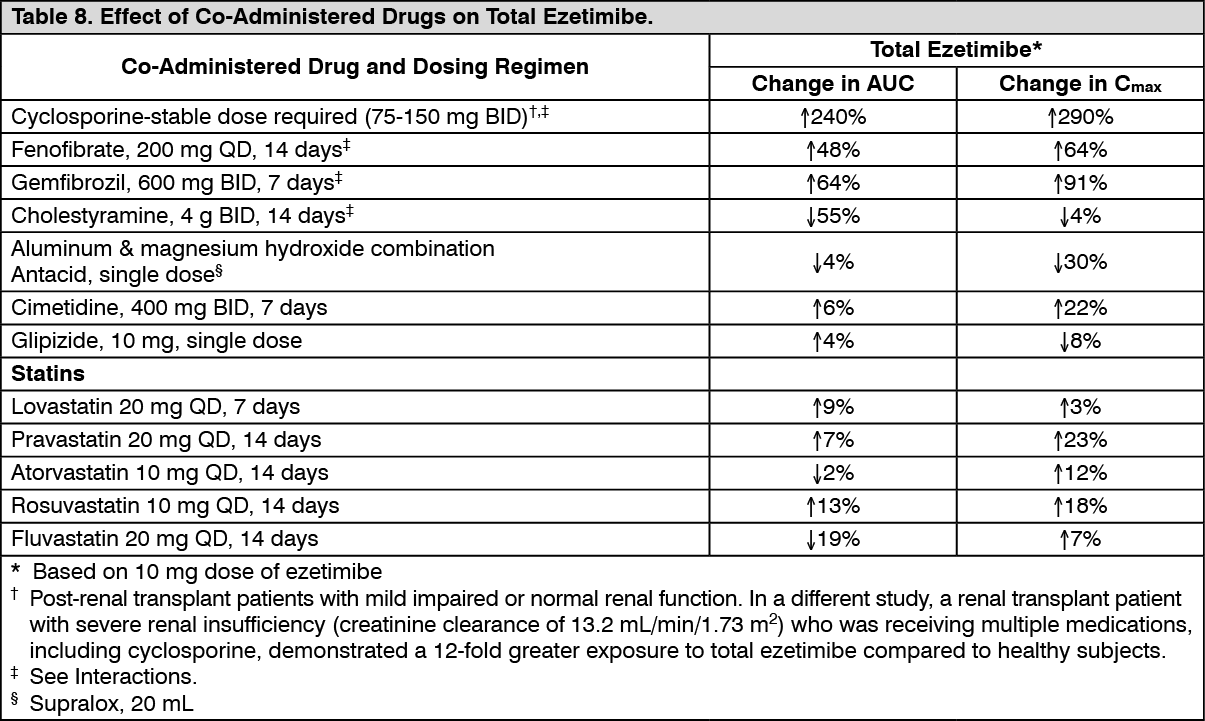 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image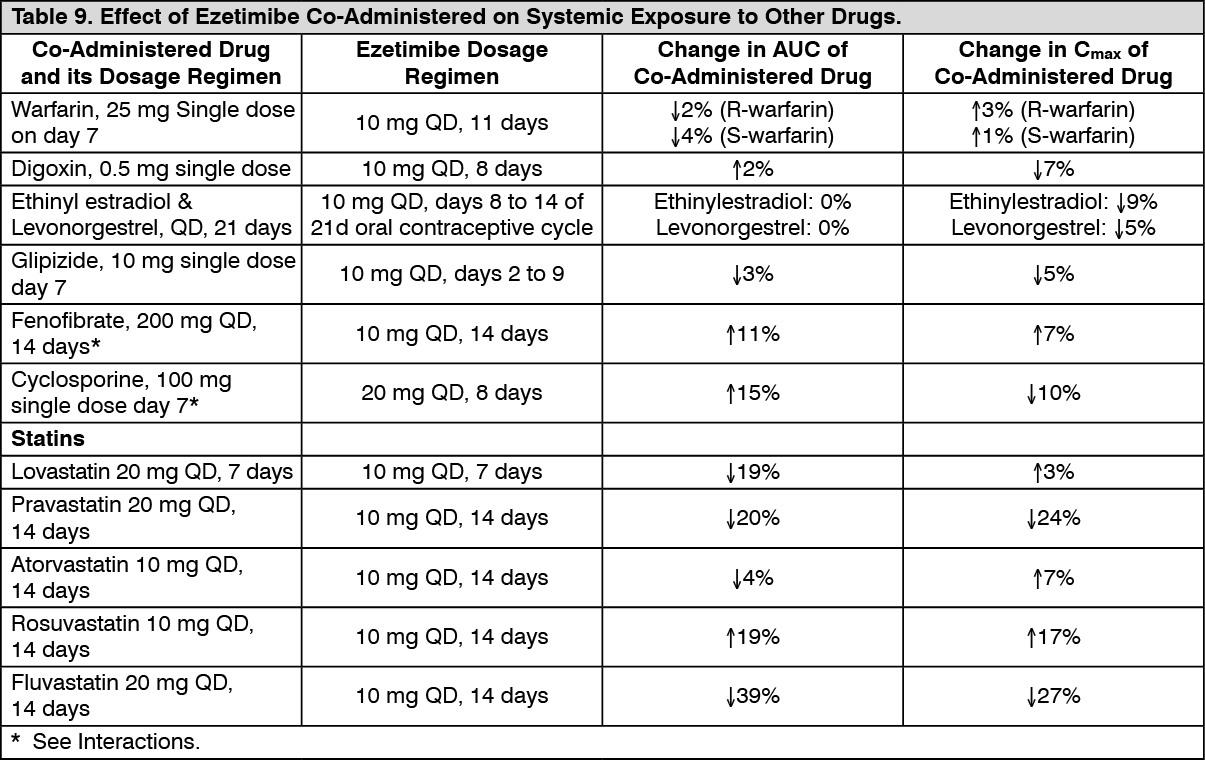 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: A 104-week dietary carcinogenicity study with ezetimibe was conducted in rats at doses up to 1,500 mg/kg/day (males) and 500 mg/kg/day (females) (~20 X the human exposure at 10 mg daily based on AUC for total ezetimibe). A 0 to 24 hr,104-week dietary carcinogenicity study with ezetimibe was also conducted in mice at doses up to 500 mg/kg/day (>150 X the human exposure at 10 mg daily based on AUC for total ezetimibe). There were no statistically significant 0 to 24 hr increases in tumor incidences in drug-treated rats or mice.
No evidence of mutagenicity was observed in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with or without metabolic activation. In addition, there was no evidence of genotoxicity in the in vivo mouse micronucleus test.
In oral (gavage) fertility studies of ezetimibe conducted in rats, there was no evidence of reproductive toxicity at doses up to 1,000 mg/kg/day in male or female rats (~7 X the human exposure at 10 mg daily based on AUC for total 0 to 24 hr ezetimibe).
Animal Toxicology and/or Pharmacology: The hypocholesterolemic effect of ezetimibe was evaluated in cholesterol-fed Rhesus monkeys, dogs, rats, and mouse models of human cholesterol metabolism. Ezetimibe was found to have an ED50 value of 0.5 mcg/kg/day for inhibiting the rise in plasma cholesterol levels in monkeys. The ED50 values in dogs, rats, and mice were 7, 30, and 700 mcg/kg/day, respectively. These results are consistent with ezetimibe being a potent cholesterol absorption inhibitor.
In a rat model, where the glucuronide metabolite of ezetimibe (SCH 60,663) was administered intraduodenally, the metabolite was as potent as the parent compound (SCH 58,235) in inhibiting the absorption of cholesterol, suggesting that the glucuronide metabolite had activity similar to the parent drug.
In 1-month studies in dogs given ezetimibe (0.03 to 300 mg/kg/day), the concentration of cholesterol in gallbladder bile increased ~2- to 4-fold. However, a dose of 300 mg/kg/day administered to dogs for one year did not result in gallstone formation or any other adverse hepatobiliary effects. In a 14-day study in mice given ezetimibe (0.3 to 5 mg/kg/day) and fed a low-fat or cholesterol-rich diet, the concentration of cholesterol in gallbladder bile was either unaffected or reduced to normal levels, respectively.
A series of acute preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of 14C-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or the fat-soluble vitamins A and D.
In 4- to 12-week toxicity studies in mice, ezetimibe did not induce cytochrome P450 drug metabolizing enzymes. In toxicity studies, a pharmacokinetic interaction of ezetimibe with statins (parents or their active hydroxy acid metabolites) was seen in rats, dogs, and rabbits.