



 Sign Out
Sign Out
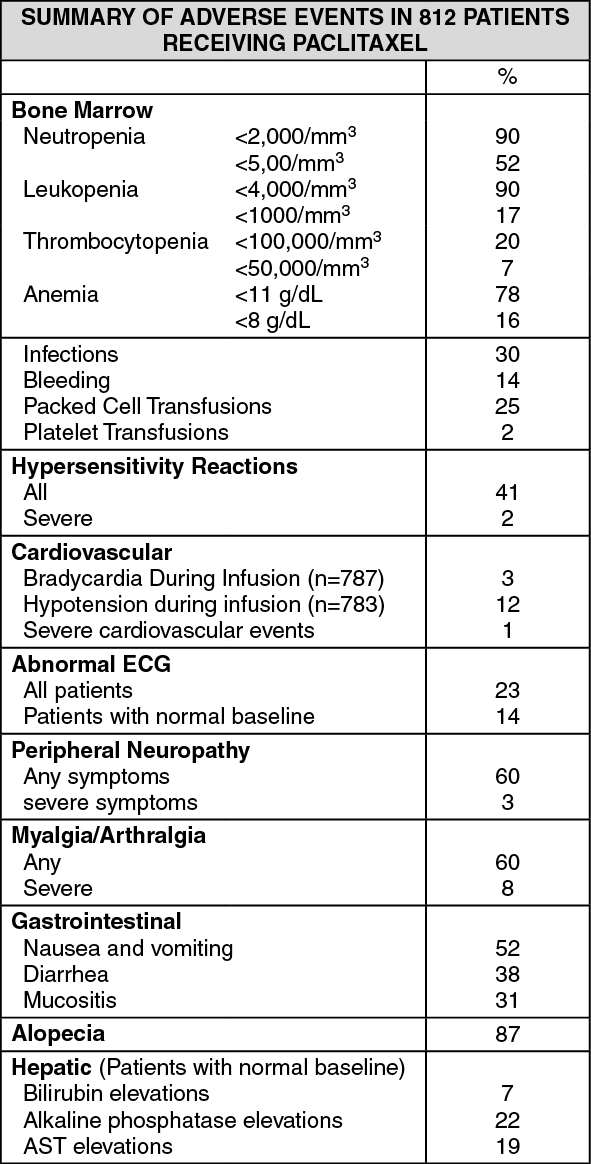 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSummary of 3 hour infusion data at dose of 175 mg/m2: Unless otherwise stated, the following safety data relate to 62 patients with ovarian cancer and 119 patients with breast cancer treated at a dose of 175 mg/m2 and a 3 hour infusion schedule, in phase III clinical trials. All patients were premedicated to minimize hypersensitivity reactions. Data from these clinical trials demonstrate that paclitaxel given at this dose and schedule is well tolerated. Myelosuppression, in particular, is less frequent and less severe than with a 24 hour infusion schedule. Moreover, as compared to a 24 hour infusion schedule, the incidence of hypersensitivity reaction, peripheral neuropathy or other significant undesirable effects was not increased when paclitaxel was administered at this dose and schedule.
None of the observed toxicities were influenced by age.
Hematologic: The most frequent significant undesirable effect of Paclitaxel was bone marrow suppression. Severe neutropenia (<500 cells/mm3) occurred in 27% of the patients, but was not associated with febrile episodes. Only 1% of the patients experienced severe neutropenia for 7 days or more.
Eighteen percent of the patients had an infectious episode. Although severe septic episodes associated with severe neutropenia attributable to Paclitaxel were reported in early clinical trials, no severe infections or septic episodes were seen at the recommended dose and infusion schedule.
Thrombocytopenia was reported in 6% of the patients. One percent of the patients had a platelet nadir count 50,000 cells/mm3 at least once while on study.
Anemia was observed in 62% of the patients, but was severe (Hb<8 g/dL) in only 6% of the patients.
Incidence and severity of anemia are related to baseline hemoglobin status.
Hypersensitivity Reactions: A significant hypersensitivity reaction (defined as hypotension requiring therapy, angioedema, respiratory distress requiring bronchodilator therapy or generalized urticaria) occurred in 2 patients. Thirty-nine percent of the patients (20% of all courses) experienced minor hypersensitivity reaction. These minor reaction, mainly flushing and rash did not require therapeutic intervention nor did they prevent continuation of Paclitaxel therapy.
Cardiovascular: Hypotension and bradycardia were experienced by 22% and 3% of the patients, respectively, and were asymptomatic in all cases.
One patient experienced transient hypertension during the second Paclitaxel cycle. In addition, 2 patients presented severe cardiovascular events (tachycardia and thrombophlebitis), but these were considered unrelated to Paclitaxel. In the same studies at a lower dose or longer infusion, 3 severe cardiovascular events (atrioventricular (AV) block, syncope and hypotension associated with coronary stenosis resulting in death) possibly related to Paclitaxel administration were reported. In early clinical studies, conducted with varying dosage and infusion schedules, 4(2%) patients experienced severe cardiovascular events possibly related to Paclitaxel which included asymptomatic ventricular tachycardia, tachycardia with bigeminy, AV block and syncope.
Neurologic: Peripheral neuropathy, mainly manifested by paresthesia, affected 64% of the patients, but was severe in only 4% of the patients. Peripheral neuropathy can occur following the first course and can worsen with increasing exposure to Paclitaxel. Sensory symptoms have usually improved or resolved within several months of Paclitaxel discontinuation. Pre-existing neuropathies resulting from prior therapies are not a contraindication for Paclitaxel therapy.
Arthralgia/Myalgia: Arthralgia or myalgia affected 54% of the patients and was severe in 12% of the patients.
Alopecia: Alopecia was observed in almost all patients.
Gastrointestinal: Gastrointestinal side effects were usually mild to moderate: nausea/vomiting, diarrhea and mucositis were reported by 44%, 25% and 20% of the patients, respectively. Other gastrointestinal events included anorexia (25% of patients), constipation (18%) and intestinal obstruction (4%).
Hepatic: Severe elevations (>5 x normal values) in AST (SGOT), alkaline phosphatase or bilirubin were seen in 5%, 5% and 1% of patients, respectively.
View ADR Reporting Link