Rosuvastatin: The adverse reactions seen with Rosuvastatin are generally mild and transient. In controlled clinical trials, less than 4% of Rosuvastatin-treated patients were withdrawn due to adverse reactions.
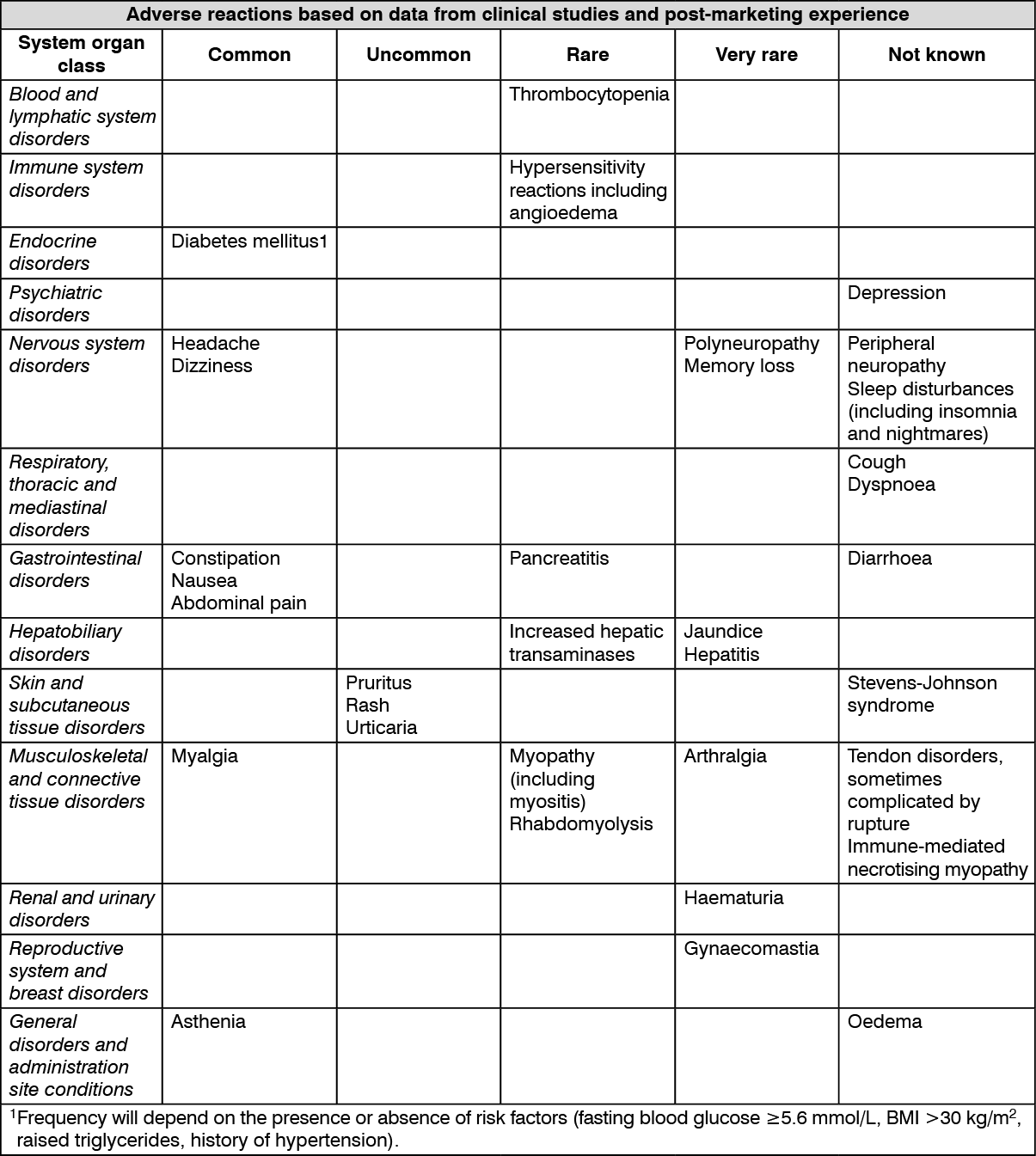
Tabulated list of adverse reactions: Based on data from clinical studies and extensive post-marketing experience, the following table presents the adverse reaction profile for rosuvastatin. Adverse reactions listed as follows are classified according to frequency and system organ class (SOC).
The frequencies of adverse reactions are ranked according to the following convention: Common (≥/100 to <1/10); Uncommon (≥1/1,000 to <1/100); Rare (≥1/10,000 to <1/1000); Very rare (<1/10,000); Not known (cannot be estimated from the available data). (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
As with other HMG-CoA reductase inhibitors, the incidence of adverse drug reactions tends to be dose dependent.
Fenofibrate: The frequencies of adverse events are ranked according top the following: Very common (> 1/10), Common (> 1/100, < 1/10), Uncommon (> 1/1,000, < 1/100), Rare (>1/10,000, < 1/1,000), very rare (< 1/10,000) including isolated reports.
Gastrointestinal: Common: Digestive, gastric or intestinal disorders (abdominal pain, nausea, vomiting, diarrhoea, and flatulence) moderate in severity.
Uncommon: Pancreatitis.
Hepato-biliary disorders: Common: Moderately elevated levels of serum transaminases (see Precautions).
Uncommon: Development of gallstones.
Very rare: Episodes of hepatitis. When symptoms (e.g. jaundice, pruritus) indicative of hepatitis occur, laboratory tests are to be conducted for verification and fenofibrate discontinued, if applicable (see Precautions).
Cardiovascular system: Uncommon: Thromboembolism (pulmonary embolism, deep vein thrombosis).
Skin and subcutaneous tissue disorder: Uncommon: rashes, pruritus, urticaria or photosensitivity reactions.
Rare: alopecia.
Very rare: cutaneous photosensitivity with erythema, vesiculation or nodulation on parts of the skin expose to sunlight or artificial light (e.g. sunlamp) in individual cases (even after many months of uncomplicated use).
Musculoskeletal, connective tissue and bone disorders: Rare: diffuse myalgia, myositis, muscular cramps and weakness.
Not known: rhabdomyolysis.
Blood and lymphatic system disorders: Rare: decrease in haemoglobin and leukocytes.
Nervous system disorder: Rare: sexual asthenia.
Respiratory, thoracic and mediastinal disorders: Not known: interstitial pneumopathies.
Investigation: Uncommon: increases in serum creatinine and urea.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out