Pharmacotherapeutic group: Lipid modifying agents, plain, HMG CoA reductase inhibitors.
ATC code: C10AA07.
Pharmacology: 10 mg: Rosuvastatin is a selective and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl coenzyme A to mevalonate, a precursor of cholesterol (total C), LDL-C, ApoB, and nonHDL-C (total cholesterol minus HDL-C) in patients with homozygous and heterozygous familial hypercholesterolemia (FM), nonfamilial forms of hypercholesterolemia, and mixed dyslipidemia. Rosuvastatin also reduces TG and produces increase in HDL-C. Rosuvastatin reduces total-C, LDL-C, VLDL-cholesterol (VLDL-C), ApoB, nonHDL-C and TG, and increases HDL-C in patients with isolated hypertriglyceridemia. The effect of rosuvastatin on cardiovascular morbidity and mortality has not been determined.
20 mg: Pharmacodynamics: Mechanism of action: Rosuvastatin is a selective and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy- 3-methylglutaryl coenzyme A to mevalonate, a precursor for cholesterol. The primary site of action rosuvastatin is the liver, the target organ for cholesterol lowering.
Rosuvastatin increases the number of hepatic LDL receptors on the cell-surface, enhancing uptake and catabolism of LDL and it inhibits the hepatic synthesis of VLDL, thereby reducing the total number of VLDL and LDL particles.
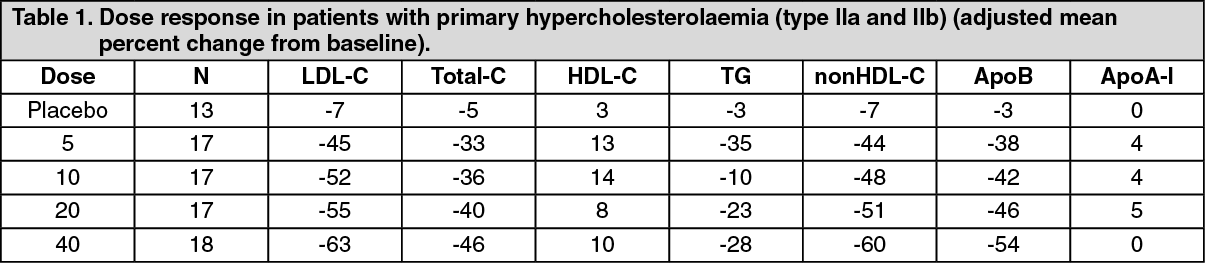
Pharmacodynamic effects: Rosuvastatin reduces elevated LDL-cholesterol, total cholesterol and triglycerides and increases HDL-cholesterol. It also lowers ApoB, nonHDL-C, VLDL-C, VLDL-TG and increases ApoA-I (see Table 1). Rosuvastatin also lowers the LDL-C/HDL-C, total C/HDL-C and nonHDL-C/HDL-C and the ApoB/ApoA-I ratios. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A therapeutic effect is obtained within 1 week following treatment initiation and 90% of maximum response is achieved in 2 weeks. The maximum response usually achieved by 4 weeks and is maintained after that.
Clinical efficacy and safety: Rosuvastatin is effective in adults with hypercholesterolaemia, with and without hypertriglyceridemia, regardless of race, sex, or age and in special populations such as diabetics, or patients with familial hypercholesterolaemia.
From pooled phase III data, rosuvastatin has been shown to be effective at treating the majority of patients with type IIa and IIb hypercholesterolaemia (mean baseline LDL-C about 4.8 mmol/L) to recognised European Atherosclerosis Society (EAS; 1998) guideline targets; about 80% of patients treated with 10 mg reached the EAS targets for LDL-C levels (<3 mmol/L).
In a large study, 435 patients with heterozygous familial hypercholesterolaemia were given rosuvastatin from 20 mg to 80 mg in a force-titration design. All doses showed a beneficial effect on lipid parameters and treatment to target goals. Following titration to a daily dose of 40 mg (12 weeks of treatment), LDL-C was reduced by 53%. Thirty-three percent (33%) of patients reached EAS guidelines for LDL-C levels (<3 mmol/L).
In a force titration, open label trial, 42 patients with homozygous familial hypercholesterolemia were evaluated for their response to rosuvastatin 20-40 mg. In the overall population, the mean LDL-C reduction was 22%.
In clinical studies with a limited number of patients, rosuvastatin has been shown to have additive efficacy in lowering triglycerides when used in combination with fenofibrate and in increasing HDL-C levels when used in combination with niacin (see Precautions).
In a multi-centre, double-blind, placebo-controlled clinical study (METEOR), 984 patients between 45 and 70 years of age and at low risk of coronary heart disease (defined as Framingham risk <10% over 10 years), with a mean LDL-C of 4.0 mmol/L (154.5 mg/dL), but with subclinical atherosclerosis (detected by Carotid Intima Media Thickness) were randomised to 40 mg rosuvastatin once daily or placebo for 2 years. Rosuvastatin significantly slowed the rate of progression of the maximum CIMT for the 12 carotid artery sites compared to placebo by -0.0145 mm/year [95% confidence interval -0.0196, -0.0093; p<0.0001]. The change from baseline was -0.0014 mm/year (-0.12%/year (non-significant)) for rosuvastatin compared to a progression of +0.0131 mm/year (1.12%/year (p<0.0001)) for placebo. No direct correlation between CIMT decrease and reduction of the risk of cardiovascular events has yet been demonstrated. The population studied in METEOR is low risk for coronary heart disease and does not represent the target population of rosuvastatin 40 mg. The 40 mg dose should only be prescribed in patients with severe hypercholesterolaemia at high cardiovascular risk (see Dosage & Administration).
In the Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) study, the effect of rosuvastatin on the occurrence of major atherosclerotic cardiovascular disease events was assessed in 17,802 men (≥50 years) and women (≥ 60 years).
Study participants were randomly assigned to placebo (n=8,901) or rosuvastatin 20 mg once daily (n=8,901) and were followed for a mean duration of 2 years.
LDL-cholesterol concentration was reduced by 45% (p<0.001) in the rosuvastatin group compared to the placebo group.
In a post-hoc analysis of a high-risk subgroup of subjects with a baseline Framingham risk score >20% (1,558 subjects), there was a significant reduction in the combined end-point of cardiovascular death, stroke and myocardial infarction (p=0.028) on rosuvastatin treatment versus placebo. The absolute risk reduction in the event rate per 1,000 patient-years was 8.8. Total mortality was unchanged in this high-risk group (p=0.193). In a post-hoc analysis of a high-risk subgroup of subjects (9,302 subjects total) with a baseline SCORE risk ≥5% (extrapolated to include subjects above 65 years), there was a significant reduction in the combined end-point of cardiovascular death, stroke and myocardial infarction (p=0.0003) on rosuvastatin treatment versus placebo. The absolute risk reduction in the event rate was 5.1 per 1,000 patient-years. Total mortality was unchanged in this high risk group (p=0.076).
In the JUPITER trial, there was 6.6% of rosuvastatin and 6.2% of placebo subjects who discontinued use of study medicinal product due to an adverse event. The most common adverse events that led to treatment discontinuation were: myalgia (0.3% rosuvastatin, 0.2% placebo), abdominal pain (0.03% rosuvastatin, 0.02% placebo) and rash (0.02% rosuvastatin, 0.03% placebo). The most common adverse events at a greater rate than or equal to placebo were urinary tract infection (8.7% rosuvastatin, 8.6% placebo), nasopharyngitis (7.6% rosuvastatin, 7.2% placebo), back pain (7.6% rosuvastatin, 6.9% placebo) and myalgia (7.6% rosuvastatin, 6.6% placebo).
Pediatric population: In a double-blind, randomised, multi-centre, placebo-controlled, 12-week study (n=176, 97 male and 79 female) followed by a 40-week (n=173, 96 male and 77 female), open-label, rosuvastatin dose-titration phase, patients 10-17 years of age (Tanner stage II-V, females at least 1 year post menarche) with heterozygous familial hypercholesterolaemia received rosuvastatin 5, 10 or 20 mg or placebo daily for 12 weeks and then all received rosuvastatin daily for 40 weeks. At study entry, approximately 30% of the patients were 10-13 years and approximately 17%, 18%, 40% and 25% were Tanner stage II, III, IV, and V, respectively.
LDL-C was reduced 38.3%, 44.6%, and 50.0% by rosuvastatin 5, 10, and 20 mg, respectively, compared to 0.7% for placebo.
At the end of the 40-week, open-label, titration to goal, dosing up to a maximum of 20 mg once daily, 70 of 173 patients (40.5%) had achieved the LDL-C goal of less than 2.8 mmol/L.
After 52 weeks of study treatment, no effect on growth, weight, BMI or sexual maturation was detected (see Precautions).
This trial (n=176) was not suited for comparison of rare adverse events.
Rosuvastatin was also studied in a 2-year open-label, titration-to-goal study in 198 children with heterozygous familial hypercholesterolaemia aged 6 to 17 years (88 male and 110 female, Tanner stage <II-V). The starting dose for all patients was 5 mg rosuvastatin once daily. Patients aged 6 to 9 years (n=64) could titrate to a maximum dose of 10 mg once daily and patients aged 10 to 17 years (n=134) to a maximum dose of 20 mg once daily.
After 24 months of treatment with rosuvastatin, the LS mean percent reduction from the baseline value in LDL-C was -43% (Baseline: 236 mg/dL, Month 24: 133 mg/dL). For each group, the LS mean percent reductions from baseline value in LDL-C were -43% (Baseline: 234 mg/dL, Month 24: 124 mg/dL), -45% (Baseline: 234 mg/dL, 124 mg/dL), and -35% (Baseline: 241 mg/dL, Month 24: 153 mg/dL) in the 6 to <10, 10 to <14, and 14 to <18 age groups, respectively.
Rosuvastatin 5 mg, 10 mg, and 20 mg also achieved statistically mean changes from baseline for the following secondary lipid and lipoprotein variables: HDL-C, TC, non-HDL-C, LDL-C/HDL-C, TC/HDL-C, TG/HDL-C, non-HDL-C/HDL-C, ApoB, ApoB/ApoA-1. These changes were each in the direction of improved lipid responses and were sustained over 2 years.
No effect on growth, weight, BMI or sexual maturation was detected after 24 months of treatment (see Precautions).
The European Medicines Agency has waived the obligation to submit the results of studies with rosuvastatin in all subsets of the paediatric population in the treatment of homozygous familial hypercholesterolaemia, primary combined (mixed) dyslipidaemia and in the prevention of cardiovascular events (see Dosage & Administration).
Pharmacokinetics: 10 mg: Absorption: The absolute bioavailability of rosuvastatin is approximately 20%. Plasma concentrations of rosuvastatin do not differ following evening or morning drug administration. Significant LDL-C reductions are seen when rosuvastatin is given with or without food, and regardless of the time of day of drug administration.
Distribution: Mean volume of distribution at steady-state of rosuvastatin is approximately 134 liters. Rosuvastatin is 88% bound to plasma proteins. This binding is reversible and independent of plasma concentrations.
Metabolism: Rosuvastatin is not extensively metabolized, approximately 10% of radiolabeled dose is recovered as metabolite. The major metabolite is N-desmethyl rosuvastatin, which is formed principally by cytochrome P450 2C9, and
in vitro studies have demonstrated that N-desmethyl rosuvastatin has approximately one-sixth to one-half the HMG-CoA reductase inhibitory activity of rosuvastatin.
Excretion: Following oral administration, rosuvastatin and its metabolites are primary excreted in the faeces (90%). The elimination half-life of rosuvastatin is approximately 19 hours.
20 mg: Absorption: Maximum rosuvastatin plasma concentrations are achieved approximately 5 hours after oral administration. The absolute bioavailability is approximately 20%.
Distribution: Rosuvastatin is taken up extensively by the liver which is the primary site of cholesterol synthesis and LDL-C clearance. The volume of distribution of rosuvastatin is approximately 134 L. Approximately 90% of rosuvastatin is bound to plasma proteins, mainly to albumin.
Biotransformation: Rosuvastatin undergoes limited metabolism (approximately 10%).
In vitro metabolism studies using human hepatocyte indicate that rosuvastatin is a poor substrate for cytochrome P450-based metabolism. CYP2C9 was the principal isoenzyme involved, with 2C19, 3A4 and 2D6 involved to a lesser extent. The main metabolites identified are the N-desmethyl and lactone metabolites. The N-desmethyl metabolite is approximately 50% less active than rosuvastatin whereas the lactone from is considered clinically inactive. Rosuvastatin accounts for greater than 90% of the circulating HMG-CoA reductase inhibitor activity.
Elimination: Approximately 90% of the rosuvastatin dose is excreted unchanged in the faeces (consisting of absorbed and non-absorbed active substance and the remaining part is excreted in urine. Approximately 5% is excreted unchanged in urine. The plasma elimination half-life is approximately 19 hours. The elimination half-life does not increase at higher doses. This geometric mean plasma clearance is approximately 50 litres/hour (coefficient of variation 21.7%). As with other HMG-CoA reductase inhibitors, the hepatic uptake of rosuvastatin involves the membrane transporter OATP-C. This transporter is important in the hepatic elimination of rosuvastatin.
Linearity: Systemic exposures of rosuvastatin increases in proportion to dose. There are no changes in pharmacokinetic parameters following multiple doses.
Special Populations: Age and sex: There was no clinically relevant effect of age or sex on the pharmacokinetics of rosuvastatin in adults. The exposure in children and adolescents with heterozygous familial hypercholesterolaemia appears to be similar to or lower than that in adult patients with dyslipidaemia (see Paediatric population as follows).
Race: Pharmacokinetic studies show an approximate 2-fold elevation in median AUC and C
max in Asian subjects (Japanese, Chinese, Filipino, Vietnamese, and Koreans) compared with Caucasians; Asian-Indians who an approximate 1.3-fold elevation in median AUC and C
max. A population pharmacokinetic analysis revealed no clinically relevant differences in pharmacokinetics between Caucasian and Black groups.
Renal impairment: In a study in subjects with varying degrees of renal impairment, mild to moderate renal disease had no influence on plasma concentration of rosuvastatin or the N-desmethyl metabolite. Subjects with severe impairment (CrCl <30 mL/min) had a 3-fold increase in plasma concentration and a 9-fold increase in the N-desmethyl metabolite concentration compared to healthy volunteers. Steady-state plasma concentration of rosuvastatin in subjects undergoing haemodialysis were approximately 50% greater compared to healthy volunteers.
Hepatic impairment: In a study with subjects with varying degrees of hepatic impairment there was no evidence of increased exposure to rosuvastatin in subjects with Child-Pugh scores of 7 or below. However, two subjects with Child- Pugh scores of 8 and 9 showed an increase in systemic exposure of at least 2-fold compared to subjects with lower Child-Pugh scores. There is no experience in subjects with Child-Pugh scores above 9.
Genetic polymorphisms: Disposition of HMG-CoA reductase inhibitors, including rosuvastatin, involves OATP1B1 and BCRP transporter proteins. In patients with SLCO1B1 (OATP1B1) and/or ABCG2 (BCRP) genetic polymorphisms, there is a risk of increased rosuvastatin exposure. Individual polymorphisms of SLCO1B1 c.521CC and ABCG2 c.421AA are associated with a higher rosuvastatin exposure (AUC) compared to SLCO1B1 c.521TT or ABCG2 c.421CC genotypes. This specific genotyping is not established in clinical practice, but for patients who are known to have these types of polymorphisms, a lower daily dose of rosuvastatin is recommended.
Paediatric population: Two pharmacokinetic studies with rosuvastatin (given as tablets) in paediatric patients with heterozygous familial hypercholesterolaemia 10-17 or 6-17 years of age (total of 214 patients) demonstrated that exposure in paediatric patients appears comparable to or lower than that in adult patients. Rosuvastatin exposure was predictable with respect to dose and time over a 2-year period.
Toxicology: Preclinical safety data: 20 mg: Preclinical data reveal no special hazard for humans based on conventional studies on safety pharmacology, genotoxicity and carcinogenicity potential. Specific tests for effects on hERG have not been evaluated. Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels were as follows: In repeated-dose toxicity studies, histopathologic liver changes likely due to the pharmacologic action of rosuvastatin were observed in mouse, rat, and to a lesser extent with effects in the gall bladder in dogs, but not in monkeys. In addition, testicular toxicity was observed in monkeys and dogs at higher doses. Reproductive toxicity was evident in rats, with reduced litter sizes, litter weight and pup survival observed at maternally toxic doses, where systemic exposures were several times above the therapeutic exposure level.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image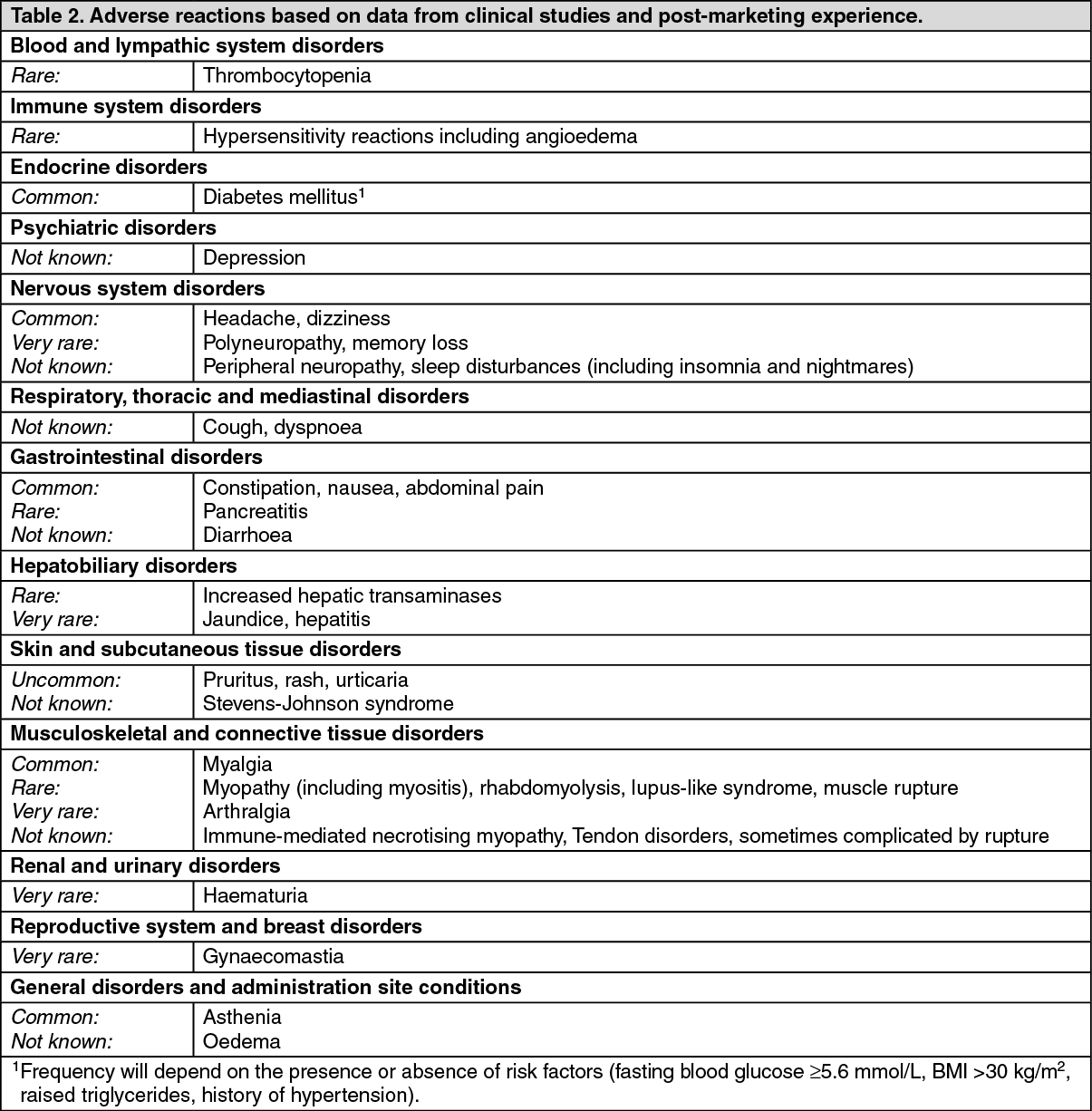 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image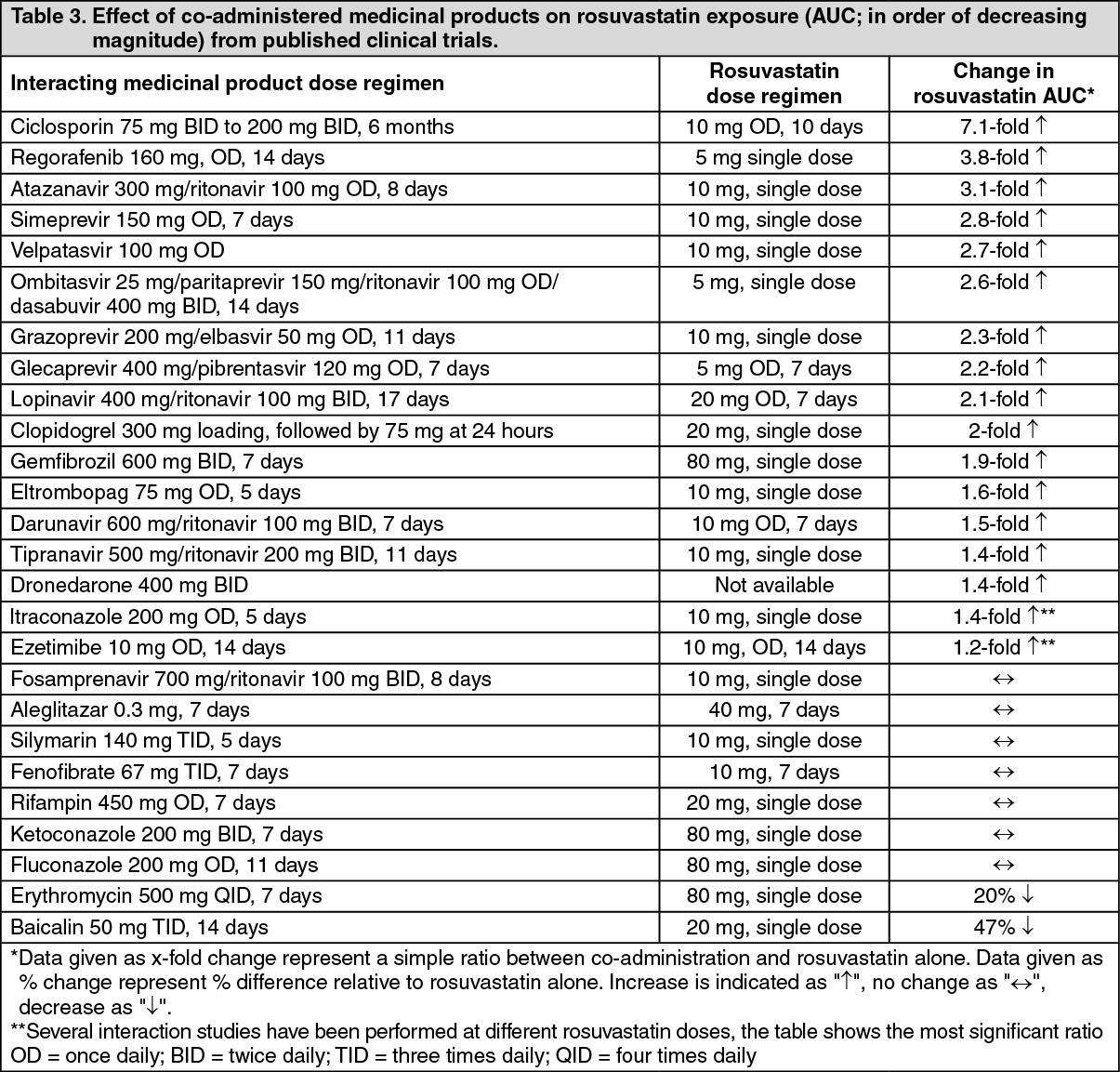 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out