Each pre-filled syringe contains 60 mg of denosumab in 1.0 mL solution (60 mg/mL).
It has pH 5.2 and may contain trace amounts of translucent to white proteinaceous particles.
Pharmacology: Pharmacodynamics: Mechanism of Action: Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and specificity to RANKL preventing RANKL from activating its only receptor, RANK, on the surface of osteoclasts and their precursors, independent of bone surface. Prevention of the RANKL/RANK interaction inhibits osteoclast formation, function, and survival. Denosumab therefore reduces bone resorption and increases bone mass and strength in both cortical and trabecular bone.
Pharmacodynamic Effects: In clinical studies, treatment with 60 mg of denosumab resulted in rapid reduction in the bone resorption marker serum type 1 C-telopeptides (CTX) within 6 hours of subcutaneous administration (by approximately 70%) with reductions of approximately 85% occurring by 3 days. CTX reductions were maintained over the 6-months dosing interval. At the end of each dosing interval, CTX reductions were partially attenuated from maximal reduction of ≥87% to approximately ≥45% (range 45-80%), reflecting the reversibility of denosumab's effects on bone remodelling once serum levels diminish. These effects were sustained with continued treatment. Consistent with the physiological coupling of bone formation and resorption in skeletal remodelling, reductions in bone formation markers [e.g. bone specific alkaline phosphatase (BSAP) and serum N-terminal propeptide of type I collagen (P1NP)] were observed beginning 1 month after the first dose of Denosumab.
Bone turnover markers (bone resorption and formation makers) generally reached pre-treatment levels within 9 months after the last 60 mg subcutaneous dose. Upon re-initiation, the degree of inhibition of CTX by Denosumab was similar to that observed in patients initiating denosumab treatment.
In a clinical study of postmenopausal women with low bone mass (N=504) who were previously treated with alendronate for a median duration of 3 years, those transitioning to receive Denosumab experienced additional reductions in serum CTX, compared with women who remained on alendronate. In this study the changes in serum calcium were similar between the two groups.
Immunogenicity: Denosumab is a human monoclonal antibody; as with all therapeutic proteins, there is a theoretical potential for immunogenicity. More than 13,000 patients were screened for binding antibodies using a sensitive electrochemiluminescent bridging immunoassay. Less than 1% of patients treated with denosumab for up to 5 years tested positive (including pre-existing, transient and developing antibodies). The patients that tested positive for binding antibodies were further evaluated for neutralising antibodies using a chemiluminescent cell-based in vitro biological assay and none of them tested positive. No evidence of altered pharmacokinetic profile, toxicity profile, or clinical response was associated with binding antibody development.
Clinical Studies: Treatment of Postmenopausal Osteoporosis: The efficacy and safety of denosumab in the treatment of postmenopausal osteoporosis was demonstrated in FREEDOM, a 3-year, randomised, double-blind, placebo-controlled, multinational study that demonstrated that denosumab was effective compared to placebo in reducing new vertebral, non-vertebral and hip fractures in post-menopausal women with osteoporosis.
7,808 women aged 60-91 years were enrolled of which 23.6% had prevalent vertebral fractures.
Women were randomised to receive subcutaneous injections of either placebo (n=3,906) or denosumab 60 mg (n=3,902) once every 6 months. Women received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily. The primary efficacy variable was the incidence of new vertebral fractures. Secondary efficacy variables included the incidence of non-vertebral fractures and hip fractures, assessed at 3 years.
Denosumab significantly reduced the risk of new vertebral, nonvertebral, and hip fractures compared with placebo. All 3 efficacy fracture endpoints achieved the statistical significance level based on the pre-specified sequential testing scheme.
Effect on Vertebral Fractures: Denosumab significantly reduced the risk of new vertebral fractures (primary endpoint) by 68% (risk ratio: 0.32; p<0.0001) over 3 years. The 3-year fracture rates for new vertebral fractures were 7.2% in the placebo group and 2.3% in the Denosumab (Prolia) group (unadjusted absolute risk reduction of 4.8%). Reductions were also observed over 1 year (61% relative risk reduction; 1.4% unadjusted absolute risk reduction) and 2 years (71% relative risk reduction; 3.5% unadjusted absolute risk reduction) (all p<0.0001).
Denosumab also reduced the risk of other prespecified categories of fractures, including new and worsening vertebral fractures (67% relative risk, reduction, 4.8% unadjusted absolute risk reduction), multiple new vertebral fractures (61% relative risk reduction, 1.0% unadjusted absolute risk reduction), clinical vertebral fractures (69% relative risk reduction, 1.8% unadjusted absolute risk reduction) over 3 years.
The reductions in the risk of new vertebral fractures by denosumab over 3 years were consistent and significant regardless of 10-year major osteoporotic baseline fracture risk as assessed by FRAX (WHO's Fracture Risk Assessment Tool algorithm) and whether or not women had a prevalent vertebral fracture or history of a non-vertebral fracture, and regardless of baseline age, BMD, bone turnover level and prior use of a medicinal product for osteoporosis.
In postmenopausal women with osteoporosis over the age of 75, Denosumab reduced the incidence of new vertebral (64%), and non-vertebral (16%) fractures.
Effect on All Clinical Fractures: Denosumab significantly decreased the risk of non-vertebral fractures (secondary endpoint) by 20% (hazard ratio: 0.80; p=0.0106) over 3 years. Three-year non-vertebral fracture rates were 8.0% in the placebo group to 6.5% in the denosumab group (unadjusted absolute risk reduction of 1.5%).
Denosumab also reduced the risk of clinical (30% relative risk reduction, 2.9% unadjusted absolute risk reduction), major non-vertebral (20% relative risk reduction, 1.2% unadjusted absolute risk reduction), and major osteoporotic fractures (35% relative risk reduction, 2.7% unadjusted absolute risk reduction) over 3 years.
In women with baseline femoral neck BMD T-score ≤-2.5, denosumab reduced the incidence of non-vertebral fractures (35% relative risk reduction, 4.1% unadjusted absolute risk reduction, p<0.001) over 3 years. Reductions in non-vertebral fractures were observed regardless of baseline 10-year probability of a major osteoporotic fracture as assessed by FRAX.
Effect on Hip Fractures: Denosumab significantly decreased the risk of hip fractures (secondary endpoint) by 40% (hazard ratio: 0.60; p=0.0362) over 3 years. Three-year hip fracture rates were 1.2% in the placebo group and 0.7% in the denosumab group (unadjusted absolute risk reduction of 0.5%). The reductions in the risk of hip fractures over 3 years were consistent and significant regardless of baseline 10-year probability of a hip fracture as assessed by FRAX.
In women with high fracture risk as defined above by baseline age, BMD and prevalent vertebral fracture, a 48% relative risk reduction was observed with denosumab (1.1% unadjusted absolute risk reduction).
In a post-hoc analysis in postmenopausal women with osteoporosis over the age of 75 denosumab reduced the incidence of hip fractures (62%).
Effect on Bone Mineral Density (BMD): Denosumab significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 1, 2 and 3 years. Denosumab increased BMD by 9.2% at the lumbar spine, 6.0% at the total hip, 4.8% at the femoral neck, 7.9% at the hip trochanter, 3.5% at the distal 1/3 radius and 4.1% at the total body over 3 years. Increases in BMD at lumbar spine, total hip and hip trochanter were observed as early as 1 month after the initial dose. Denosumab increased lumbar spine BMD from baseline in 96% of postmenopausal women at 3 years. Consistent effects on BMD were observed at the lumbar spine regardless of baseline age, race, weight/BMI, BMD and bone turnover level.
Bone Histology: Histology assessments showed bone of normal architecture and quality, as well as the expected decrease in bone turnover relative to placebo treatment. There was no evidence of mineralisation defects, woven bone or marrow fibrosis.
Open-Label Extension Study in the Treatment of Postmenopausal Osteoporosis: A total of 4550 patients who completed the FREEDOM study (N=7808) enrolled in a 7-year, multinational, multicenter, open label, single-arm extension study to evaluate the long-term safety and efficacy of Denosumab (Prolia). All patients in the extension study received Denosumab every 6 months as a single 60 mg SC dose, as well as daily calcium (at least 1 g) and vitamin D (at least 400 IU).
Based on data from the first 2 years of the extension study for patients who received Denosumab in the FREEDOM study and continued on therapy (years 4 and 5 of Denosumab treatment), the overall subject incidence rates of adverse events and serious adverse events reported were similar to that observed in the initial 3 years of the FREEDOM study.
For patients who crossed over to Denosumab from placebo in the FREEDOM study, the overall subject incidence rates of adverse events and serious adverse events reported also similar to the first 3 years of the FREEDOM study. Two cases of ONJ were observed; both resolved.
Denosumab treatment maintained a low incidence of new vertebral and non-vertebral fractures in years 4 and 5 (2.8% of patients had at least one new vertebral fracture by month 24, 2.5% of patients had a nonvertebral fracture).
Denosumab treatment continued to increase BMD at the lumbar spine (3.3%), total hip (1.3%), femoral neck (1.2%) and trochanter (1.8%) in years 4 and 5. Percent increase in BMD from the original FREEDOM study baseline (ie, after 5 years of treatment) in the long-term group was 13.8% at the lumbar spine, 7.0% at the total hip, 6.2% at the femoral neck and 9.7% at the trochanter.
Comparative Clinical Data vs Alendronate in the Treatment of Postmenopausal Women with Low Bone Mass: In two randomised, double-blind, active-controlled studies, one in treatment-naive women and another in women previously treated with alendronate, Denosumab showed significantly greater increases in BMD and reductions in bone turnover markers (e.g. serum CTX), compared to alendronate.
Consistently greater increases in BMD were seen at the lumbar spine, total hip, femoral neck, hip trochanter, and distal 1/3 radius in women treated with denosumab, compared to those who continued to receive alendronate therapy (all p<0.05).
Clinical Efficacy in the Treatment of Bone Loss Associated with Hormone Ablation: Treatment of Bone Loss Associated with Androgen Deprivation: The efficacy and safety of denosumab in the treatment of bone loss associated with androgen deprivation was assessed in a 3-year randomised, double-blind, placebo-controlled, multinational study of 1,468 men with non-metastatic prostate cancer aged 48-97 years. Men less than 70 years of age also had either a BMD T-score at the lumbar spine, total hip, or femoral neck <-1.0 or a history of an osteoporotic fracture. Subjects either received subcutaneous injections of either denosumab 60 mg (n=734) or placebo (n=734) once every 6 months. Men received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Significant increases in BMD were observed at the lumbar spine, total hip, femoral neck and the hip trochanter as early as 1 month after the initial dose. Denosumab increased lumbar spine BMD by 7.9%, total hip BMD by 5.7%, femoral neck BMD by 4.9%, hip trochanter BMD by 6.9%, distal 1/3 radius BMD by 6.9%, and total body BMD by 4.7% over 3 years, relative to placebo (p<0.0001). Consistent effects on BMD were observed at the lumbar spine regardless of age, race, geographical region, weight/BMI, BMD, bone turnover level; duration of androgen deprivation and presence of vertebral fracture at baseline.
Denosumab significantly decreased the risk of new vertebral fractures by 62% (hazard ratio: 0.38; p<0.0063) over 3 years. Reductions were also observed over 1 year (85% relative risk reduction; 1.6% absolute risk reduction), and 2 years (69% relative risk reduction; 2.2% absolute risk reduction) (all p<0.01). Denosumab also reduced the subject incidence of more than one osteoporotic fracture at any site by 72% relative to placebo over 3 years (placebo 2.5% vs. Denosumab 0.7%, p=0.0063).
Treatment of Bone Loss in Women Undergoing Aromatase Inhibitor Therapy for Breast Cancer: The efficacy and safety of denosumab in the treatment of bone loss associated with adjuvant aromatase inhibitor therapy was assessed in a 2-year, randomised, double-blind, placebo-controlled multinational study of 252 women with non-metastatic breast cancer aged 35-84 years. Women had baseline BMD T-scores between -1.0 to -2.5 at the lumbar spine, total hip or femoral neck. Women were randomised to receive subcutaneous injections of either denosumab 60 mg (n=127) or placebo (n=125) once every 6 months. Women received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily. The primary efficacy variable was percent change in lumbar spine BMD.
Denosumab significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 2 years: 7.6% at the lumbar spine, 4.7% at the total hip, 3.6% at the femoral neck, 5.9% at the hip trochanter, 6.1% at the distal 1/3 radius and 4.2% at the total body. Significant increases in BMD were observed at the lumbar spine as early as 1 month after the initial dose. Consistent effects on BMD were observed at the lumbar spine regardless of baseline age, duration of aromatase inhibitor therapy, weight/BMI, prior chemotherapy, prior selective estrogen receptor modulator (SERM) use, and time since menopause.
Pharmacokinetics: Following subcutaneous administration, Denosumab displayed non-linear pharmacokinetics with dose over a wide dose range, and dose-proportional increases in exposure for doses of 60 mg (or 1 mg/kg) and higher.
Absorption: Following a 60 mg subcutaneous dose of Denosumab, bioavailability was 61% and maximum serum Denosumab concentrations (Cmax) of 6 μg/mL (range 1-17 μg/mL) occurred in 10 days (range 2-28 days). After Cmax, serum levels declined with a half-life of 26 days (range 6-52 days) over a period of 3 months (range 1.5-4.5 months). Fifty-three percent of patients had no measurable amounts of Denosumab detected at 6 months post-dose.
Distribution: No accumulation or change in Denosumab pharmacokinetics with time was observed upon multiple-dosing of 60 mg subcutaneously once every 6 months.
Metabolism: Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin. Based on nonclinical data, Denosumab metabolism is expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination: Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is not expected to be eliminated via hepatic metabolic mechanisms [e.g. cytochrome p450 (CYP) enzymes]. Based on nonclinical data, its elimination is expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Drug Interactions: In a study of 17 postmenopausal women with osteoporosis, midazolam (2 mg oral) was administered two weeks after a single dose of denosumab (60 mg subcutaneously), which corresponds to time of maximal pharmacodynamic effects of denosumab. Denosumab did not affect the pharmacokinetics of midazolam, which is metabolized by cytochrome P450 3A4 (CYP3A4). This indicates that denosumab should not alter the PK of drugs metabolized by CYP3A4.
Special Patient Populations: Elderly (greater than or equal to 65 Years of Age): Age was not found to be a significant factor on Denosumab pharmacokinetics in a population pharmacokinetic analysis of patients ranging in age from 28 to 87 years of age.
Children and Adolescents (up to 18 Years): No pharmacokinetic data are available in paediatric patients.
Race: The pharmacokinetics of Denosumab were not affected by race in post-menopausal women or in breast cancer patients undergoing hormone ablation.
Renal Impairment: In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics and pharmacodynamics of Denosumab; therefore dose adjustment for renal impairment is not necessary.
Hepatic Impairment: No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of Denosumab.
Toxicology: Pre-Clinical Safety Data: Carcinogenicity: The carcinogenic potential of Denosumab has not been evaluated in long-term animal studies.
Mutagenicity: The genotoxic potential of Denosumab has not been evaluated.
Reproductive Toxicology: Fertility: Denosumab had no effect on female fertility or male reproductive organs in monkeys at AUC exposures that were 100- to 150-fold higher than the human exposure at 60 mg administered subcutaneously once every 6 months.
Animal Pharmacology: Long-term treatment (16 months) of aged ovariectomized monkeys with Denosumab at doses of 25 or 50 mg/kg SC once monthly was associated with significant gains in the mass, density (BMD), and strength of cancellous and cortical bone. Bone tissue was normal with no evidence of mineralization defects, accumulation of osteoid or woven bone.
Transition from 6-months treatment with alendronate to 25 mg/kg Denosumab in ovariectomized monkeys did not cause any meaningful decreases of serum calcium. Bone strength and reduction in bone resorption at all skeletal sites were maintained or improved.
Abnormal growth plates were observed in adolescent monkeys dosed with Denosumab at 10 and 50 mg/kg SC (27 and 150 times the AUC exposure in adult humans dosed with Denosumab at 60 mg SC every 6 months), consistent with the pharmacological activity of Denosumab.
In neonatal cynomolgus monkeys exposed in utero to denosumab at 50 mg/kg, there was increased postnatal mortality; abnormal bone growth resulting in reduced bone strength, reduced haematopoiesis, and tooth malalignment; absence of peripheral lymph nodes; and decreased neonatal growth. Following a recovery period from birth to 6 months of age, the effects on bone returned to normal; there were no adverse effects on tooth eruption; and minimal to moderate mineralisation in multiple tissues was seen in one recovery animal. Maternal mammary gland development was normal. Additional information on the pharmacodynamic properties of Denosumab has been obtained from knockout mice lacking RANK or RANKL, and by the use of inhibitors of the RANKL pathway in rodents such as OPG-Fc. Knockout mice: (1) had an absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy); (2) exhibited impairment of lymph node formation; and (3) exhibited reduced bone growth and lack of tooth eruption. Similar phenotypic changes were seen in a corroborative study in 2-week old rats given OPG-Fc.
Tissue distribution studies indicated that denosumab does not bind to tissues known for expression of other members of TNF superfamily, including TNF-related apoptosis-inducing ligand (TRAIL).
Treatment of Postmenopausal Women with Osteoporosis at High Risk for Fracture: Denosumab (Prolia) is indicated for the treatment of postmenopausal women with osteoporosis at high risk for fracture, defined as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy. In postmenopausal women with osteoporosis, denosumab (Prolia) reduces the incidence of vertebral, nonvertebral, and hip fractures [see Pharmacology: Pharmacodynamics: Clinical Studies: Postmenopausal Women with Osteoporosis under Actions].
Treatment to Increase Bone Mass in Men with Osteoporosis: Denosumab (Prolia) is indicated for treatment to increase bone mass in men with osteoporosis at high risk for fracture, defined as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy [see Pharmacology: Pharmacodynamics: Clinical Studies: Treatment to Increase Bone Mass in Men with Osteoporosis under Actions].
Treatment of Glucocorticoid-Induced Osteoporosis: Denosumab (Prolia) is indicated for the treatment of glucocorticoid-induced osteoporosis in men and women at high risk of fracture who are either initiating or continuing systemic glucocorticoids in a daily dosage equivalent to 7.5 mg or greater of prednisone and expected to remain on glucocorticoids for at least 6 months. High risk of fracture is defined as a history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy [see Pharmacology: Pharmacodynamics: Clinical Studies: Treatment of Glucocorticoid-Induced Osteoporosis under Actions].
Treatment of Bone Loss in Men Receiving Androgen Deprivation Therapy for Prostate Cancer: Denosumab (Prolia) is indicated as a treatment to increase bone mass in men at high risk for fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer. In these patients denosumab (Prolia) also reduced the incidence of vertebral fractures [see Pharmacology: Pharmacodynamics: Clinical Studies: Treatment of Bone Loss in Men with Prostate Cancer under Actions].
Treatment of Bone Loss in Women Receiving Adjuvant Aromatase Inhibitor Therapy for Breast Cancer: Denosumab (Prolia) is indicated as a treatment to increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer [see Pharmacology: Pharmacodynamics: Clinical Studies: Treatment of Bone Loss in Women with Breast Cancer under Actions].
Information Essential to Safe Dosing or Administration: Pregnancy must be ruled out prior to administration of denosumab (Prolia). Perform pregnancy testing in all females of reproductive potential prior to administration of denosumab (Prolia). Based on findings in animals, denosumab (Prolia) can cause fetal harm when administered to pregnant women [see Use in Pregnancy & Lactation].
Recommended Dosage: Denosumab (Prolia) should be administered by a healthcare professional.
The recommended dose of denosumab (Prolia) is 60 mg administered as a single subcutaneous injection once every 6 months. Administer denosumab (Prolia) via subcutaneous injection in the upper arm, the upper thigh, or the abdomen. All patients should receive calcium 1000 mg daily and at least 400 IU vitamin D daily [see Hypocalcemia and Mineral Metabolism under Precautions].
If a dose of denosumab (Prolia) is missed, administer the injection as soon as the patient is available. Thereafter, schedule injections every 6 months from the date of the last injection.
Preparation and Administration: Visually inspect denosumab (Prolia) for particulate matter and discoloration prior to administration whenever solution and container permit. Denosumab (Prolia) is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter.
Latex Allergy: People sensitive to latex should not handle the grey needle cap on the single-use prefilled syringe, which contains dry natural rubber (a derivative of latex).
Prior to administration, denosumab (Prolia) may be removed from the refrigerator and brought to room temperature (up to 25°C) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm denosumab (Prolia) in any other way.
Instructions for Prefilled Syringe with Needle Safety Guard: IMPORTANT: In order to minimize accidental needlesticks, the denosumab (Prolia) single-use prefilled syringe will have a green safety guard; manually activate the safety guard after the injection is given. DO NOT slide the green safety guard forward over the needle before administering the injection; it will lock in place and prevent injection.
Activate the green safety guard (slide over the needle) after the injection.
The grey needle cap on the single-use prefilled syringe contains dry natural rubber (a derivative of latex); people sensitive to latex should not handle the cap.
Step 1: Remove Grey Needle Cap: Remove needle cap.
Step 2: Administer Subcutaneous Injection: Choose an injection site. The recommended injection sites for denosumab (Prolia) include: the upper arm OR the upper thigh OR the abdomen.
Insert needle and inject all the liquid subcutaneously. Do not administer into muscle or blood vessel.
DO NOT put grey needle cap back on needle.
Step 3: Immediately Slide Green Safety Guard Over Needle: With the needle pointing away from you.
Hold the prefilled syringe by the clear plastic finger grip with one hand. Then, with the other hand, grasp the green safety guard by its base and gently slide it towards the needle until the green safety guard locks securely in place and/or you hear a "click". DO NOT grip the green safety guard too firmly - it will move easily if you hold and slide it gently.
Hold clear finger grip.
Gently slide green safety guard over needle and lock securely in place. Do not grip green safety guard too firmly when sliding over needle.
Immediately dispose of the syringe and needle cap in the nearest sharps container. DO NOT put the needle cap back on the used syringe.
No data from clinical trials are available regarding overdosage of Denosumab (Prolia).
Denosumab has been administered in clinical studies using doses up to 180 mg every 4 weeks (cumulative doses up to 1080 mg over 6 months).
Denosumab (Prolia) is contraindicated in: Hypocalcemia: Pre-existing hypocalcemia must be corrected prior to initiating therapy with denosumab (Prolia) [see Hypocalcemia and Mineral Metabolism under Precautions].
Pregnancy: Denosumab (Prolia) may cause fetal harm when administered to a pregnant woman. In women of reproductive potential, pregnancy testing should be performed prior to initiating treatment with denosumab (Prolia) [see Use in Pregnancy & Lactation].
Hypersensitivity: Denosumab (Prolia) is contraindicated in patients with a history of systemic hypersensitivity to any component of the product. Reactions have included anaphylaxis, facial swelling, and urticaria [see Hypersensitivity under Precautions and Postmarketing Experience under Adverse Reactions].
Drug Products with Same Active Ingredient: Denosumab (Prolia) contains the same active ingredient (denosumab) found in denosumab (Xgeva). Patients receiving denosumab (Prolia) should not receive denosumab (Xgeva).
Hypersensitivity: Clinically significant hypersensitivity including anaphylaxis has been reported with denosumab (Prolia). Symptoms have included hypotension, dyspnea, throat tightness, facial and upper airway edema, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue further use of denosumab (Prolia) [see Contraindications and Postmarketing Experience under Adverse Reactions].
Hypocalcemia and Mineral Metabolism: Hypocalcemia may be exacerbated by the use of denosumab (Prolia). Pre-existing hypocalcemia must be corrected prior to initiating therapy with denosumab (Prolia). In patients predisposed to hypocalcemia and disturbances of mineral metabolism (e.g. history of hypoparathyroidism, thyroid surgery, parathyroid surgery, malabsorption syndromes, excision of small intestine, severe renal impairment [creatinine clearance < 30 mL/min] or receiving dialysis), clinical monitoring of calcium and mineral levels (phosphorus and magnesium) is highly recommended within 14 days of denosumab (Prolia) injection. In some postmarketing cases, hypocalcemia persisted for weeks or months and required frequent monitoring and intravenous and/or oral calcium replacement, with or without vitamin D.
Hypocalcemia following denosumab (Prolia) administration is a significant risk in patients with severe renal impairment [creatinine clearance < 30 mL/min] or receiving dialysis. These patients may also develop marked elevations of serum parathyroid hormone (PTH). Instruct all patients with severe renal impairment, including those receiving dialysis, about the symptoms of hypocalcemia and the importance of maintaining calcium levels with adequate calcium and vitamin D supplementation.
Adequately supplement all patients with calcium and vitamin D [see Information Essential to Safe Dosing or Administration under Dosage & Administration, Contraindications, and Clinical Trials Experience under Adverse Reactions].
Osteonecrosis of the Jaw: Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing. ONJ has been reported in patients receiving denosumab [see Clinical Trials Experience under Adverse Reactions]. A routine oral exam should be performed by the prescriber prior to initiation of denosumab (Prolia) treatment. A dental examination with appropriate preventive dentistry is recommended prior to treatment with denosumab (Prolia) in patients with risk factors for ONJ such as invasive dental procedures (e.g. tooth extraction, dental implants, oral surgery), diagnosis of cancer, concomitant therapies (e.g. chemotherapy, corticosteroids, angiogenesis inhibitors), poor oral hygiene, and comorbid disorders (e.g. periodontal and/or other pre-existing dental disease, anemia, coagulopathy, infection, ill-fitting dentures). Good oral hygiene practices should be maintained during treatment with denosumab (Prolia). Concomitant administration of drugs associated with ONJ may increase the risk of developing ONJ. The risk of ONJ may increase with duration of exposure to denosumab (Prolia).
For patients requiring invasive dental procedures, clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on individual benefit-risk assessment.
Patients who are suspected of having or who develop ONJ while on denosumab (Prolia) should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Discontinuation of denosumab (Prolia) therapy should be considered based on individual benefit-risk assessment.
Atypical Subtrochanteric and Diaphyseal Femoral Fractures: Atypical low energy or low trauma fractures of the shaft have been reported in patients receiving denosumab (Prolia) [see Clinical Trials Experience under Adverse Reactions]. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Causality has not been established as these fractures also occur in osteoporotic patients who have not been treated with antiresorptive agents. Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral, and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During denosumab (Prolia) treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of denosumab (Prolia) therapy should be considered, pending a benefit-risk assessment, on an individual basis.
Multiple Vertebral Fractures (MVF) Following Discontinuation of denosumab (Prolia) Treatment: Following discontinuation of denosumab (Prolia) treatment, fracture risk increases, including the risk of multiple vertebral fractures. Cessation of denosumab (Prolia) treatment results in markers of bone resorption increasing above pretreatment values then returning to pretreatment values 24 months after the last dose of denosumab (Prolia). In addition, bone mineral density returns to pretreatment values within 18 months after the last injection [see Postmenopausal Women with Osteoporosis as follows].
New vertebral fractures occurred as early as 7 months (on average 19 months) after the last dose of denosumab (Prolia). Prior vertebral fracture was a predictor of multiple vertebral fractures after denosumab (Prolia) discontinuation. Evaluate an individual's benefit-risk before initiating treatment with denosumab (Prolia).
If denosumab (Prolia) treatment is discontinued, consider transitioning to an alternative antiresorptive therapy [see Clinical Trials Experience under Adverse Reactions].
Serious Infections: In a clinical trial of over 7800 women with postmenopausal osteoporosis, serious infections leading to hospitalization were reported more frequently in the denosumab (Prolia) group than in the placebo group [see Clinical Trials Experience under Adverse Reactions]. Serious skin infections, as well as infections of the abdomen, urinary tract, and ear, were more frequent in patients treated with denosumab (Prolia). Endocarditis was also reported more frequently in denosumab (Prolia)-treated patients. The incidence of opportunistic infections was similar between placebo and denosumab (Prolia) groups, and the overall incidence of infections was similar between the treatment groups. Advise patients to seek prompt medical attention if they develop signs or symptoms of severe infection, including cellulitis.
Patients on concomitant immunosuppressant agents or with impaired immune systems may be at increased risk for serious infections. Consider the benefit-risk profile in such patients before treating with denosumab (Prolia). In patients who develop serious infections while on denosumab (Prolia), prescribers should assess the need for continued denosumab (Prolia®) therapy.
Dermatologic Adverse Reactions: In a large clinical trial of over 7800 women with postmenopausal osteoporosis, epidermal and dermal adverse events such as dermatitis, eczema, and rashes occurred at a significantly higher rate in the denosumab (Prolia) group compared to the placebo group. Most of these events were not specific to the injection site [see Clinical Trials Experience under Adverse Reactions]. Consider discontinuing denosumab (Prolia) if severe symptoms develop.
Musculoskeletal Pain: In postmarketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking denosumab (Prolia) [see Postmarketing Experience under Adverse Reactions]. The time to onset of symptoms varied from one day to several months after starting denosumab (Prolia). Consider discontinuing use if severe symptoms develop.
Suppression of Bone Turnover: In clinical trials in women with postmenopausal osteoporosis, treatment with denosumab (Prolia) resulted in significant suppression of bone remodeling as evidenced by markers of bone turnover and bone histomorphometry [see Postmenopausal Women with Osteoporosis as follows]. The significance of these findings and the effect of long-term treatment with denosumab (Prolia) are unknown. The long-term consequences of the degree of suppression of bone remodeling observed with denosumab (Prolia) may contribute to adverse outcomes such as osteonecrosis of the jaw, atypical fractures, and delayed fracture healing. Monitor patients for these consequences.
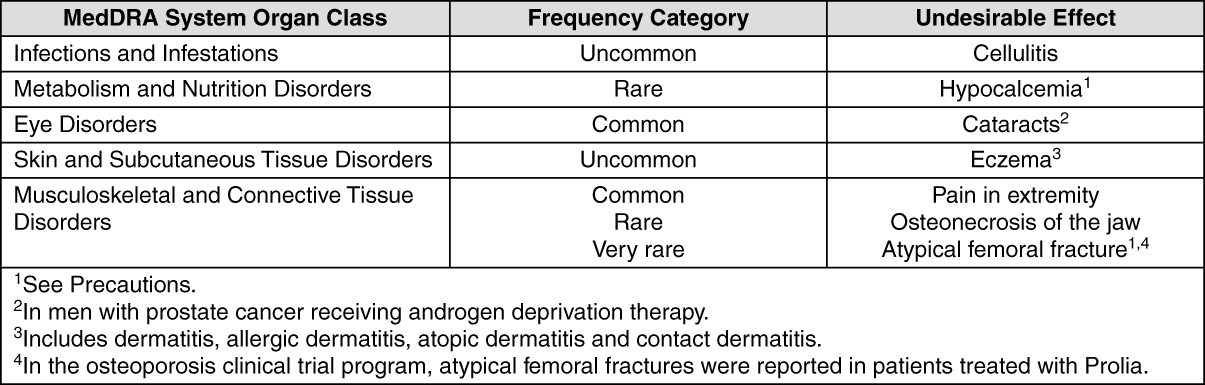
Clinical Trial Data: Adverse reactions are listed as follows by MedDRA body system organ class and by frequency. The frequency categories based on one year event rates used are: Very common ≥1 in 10; common ≥1 in 100 and <1 in 10; uncommon ≥1 in 1,000 and <1 in 100; rare ≥1 in 10,000 and <1 in 1,000; very rare <1 in 10,000.
Within each frequency grouping and system organ class, undesirable effects are presented in order of decreasing seriousness. (See table.)
 Click on icon to see table/diagram/image
Post-Marketing Data: Hypersensitivity Reactions:
Click on icon to see table/diagram/image
Post-Marketing Data: Hypersensitivity Reactions: Hypersensitivity reactions, including rash, urticaria, facial swelling, erythema and anaphylactic reactions have been reported in patients receiving Denosumab (Prolia).
Severe Hypocalcaemia: Severe symptomatic hypocalcaemia has been reported in patients at increased risk of hypocalcaemia receiving Denosumab (Prolia).
Musculoskeletal Pain: Musculoskeletal pain, including severe cases, has been reported in patients receiving Denosumab (Prolia).
Denosumab (Prolia) (60 mg subcutaneously) did not affect the pharmacokinetics of midazolam, which is metabolized by cytochrome P450 3A4 (CYP3A4), indicating that it should not affect the pharmacokinetics of drugs metabolized by this enzyme (see Pharmacology: Pharmacokinetics under Actions).
Instructions for Use or Handling: Persons sensitive to latex should not handle the needle cap on the single use prefilled syringe, which contains dry natural rubber (a derivative of latex).
Before administration, the Denosumab (Prolia) solution should be inspected for particulate matter and discolouration. The solution should not be used if cloudy or discoloured.
Do not shake.
To avoid discomfort at the site of injection, allow the pre-filled syringe to reach room temperature (up to 25°C) before injecting and inject slowly. Inject the entire contents of the pre-filled syringe. Dispose of any medicinal product remaining in the pre-filled syringe.
Instruction for self-administration by subcutaneous injection is included in the package leaflet.
Any unused product or waste material should be disposed of in accordance with local requirements.
Instructions for Injecting with Denosumab (Prolia) Pre-Filled Syringe with a Manual Needle Guard: Important: In order to minimize accidental needlesticks, the Denosumab (Prolia) single-use pre-filled syringe will have a green safety guard; manually activate the safety guard after the injection is given.
Do not slide the green safety guard forward over the needle before administering the injection; it will lock in place and prevent injection.
Activate the green safety guard (slide over the needle) after the injection.
The grey needle cap on the single-use pre-filled syringe contains dry natural rubber (a derivative of latex); people sensitive to latex should not handle the cap.
Step 1: Remove grey needle cap.
Step 2: Administer injection. Insert the needle and inject all the liquid. Do not put the grey needle cap back on the needle.
Step 3: Immediately slide the green safety guard over the needle. With the needle pointed away, hold the pre-filled syringe by the clear plastic finger grip with one hand. Then, with the other hand, grasp the green safety guard by its base and gently slide it towards the needle until the green safety guard locks securely in place and/or a "click" is heard. Do not grip the green safety guard too firmly-it will move easily if held and slide it gently.
Immediately dispose of the syringe and needle cap in the nearest sharps container. Do not put the needle cap back on the used syringe.
Store in a refrigerator (2°C-8°C).
Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from direct light.
Do not shake.
If removed from the refrigerator, Denosumab (Prolia) should be kept at a controlled room temperature (up to 25°C) in the original carton and must be used within 30 days.
M05BX04 - denosumab ; Belongs to the class of other drugs affecting bone structure and mineralization. Used in the treatment of bone diseases.
Prolia soln for inj 60 mg/mL
1 mL x 1's (P18,490/prefilled syringe)




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out