Pharmacology: Pharmacodynamics: Mechanism of action: Expression of programmed cell death ligand-1 (PD-L1) protein is an adaptive immune response that helps tumours evade detection and elimination by the immune system. PD-L1 can be induced by inflammatory signals (e.g. IFN-gamma) and can be expressed on both tumour cells and tumour-associated immune cells in tumour microenvironment. PD-L1 blocks T-cell function and activation through interaction with PD-1 and CD80 (B7.1). By binding to its receptors, PD-L1 reduces cytotoxic T-cell activity, proliferation, and cytokine production.
Durvalumab is a fully human, high affinity, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1) while leaving PD-1/PD-L2 interaction intact. Durvalumab does not induce antibody dependent cell-mediated cytotoxicity (ADCC). Selective blockade of PD-L1/PD-1 and PD-L1/CD80 interactions enhances antitumour immune responses. These antitumour responses may result in tumour elimination.
In preclinical studies, PD-L1 blockade led to increased T-cell activation and decreased tumour size.
The combination of durvalumab, a PD-L1 inhibitor, and tremelimumab, a CTLA-4 inhibitor functions to enhance anti-tumour T-cell activation and function at multiple stages of the immune response, maximizing anti-tumour immunity.
Clinical efficacy and safety: Durvalumab doses of 10 mg/kg every 2 weeks or 1500 mg every 4 weeks were evaluated in UC, NSCLC and ES-SCLC clinical studies. Based on the modelling and simulation of exposure, exposure-safety relationships and exposure-efficacy data comparisons, there are no anticipated clinically significant differences in efficacy and safety between durvalumab doses of 10 mg/kg every 2 weeks or 1500 mg every 4 weeks.
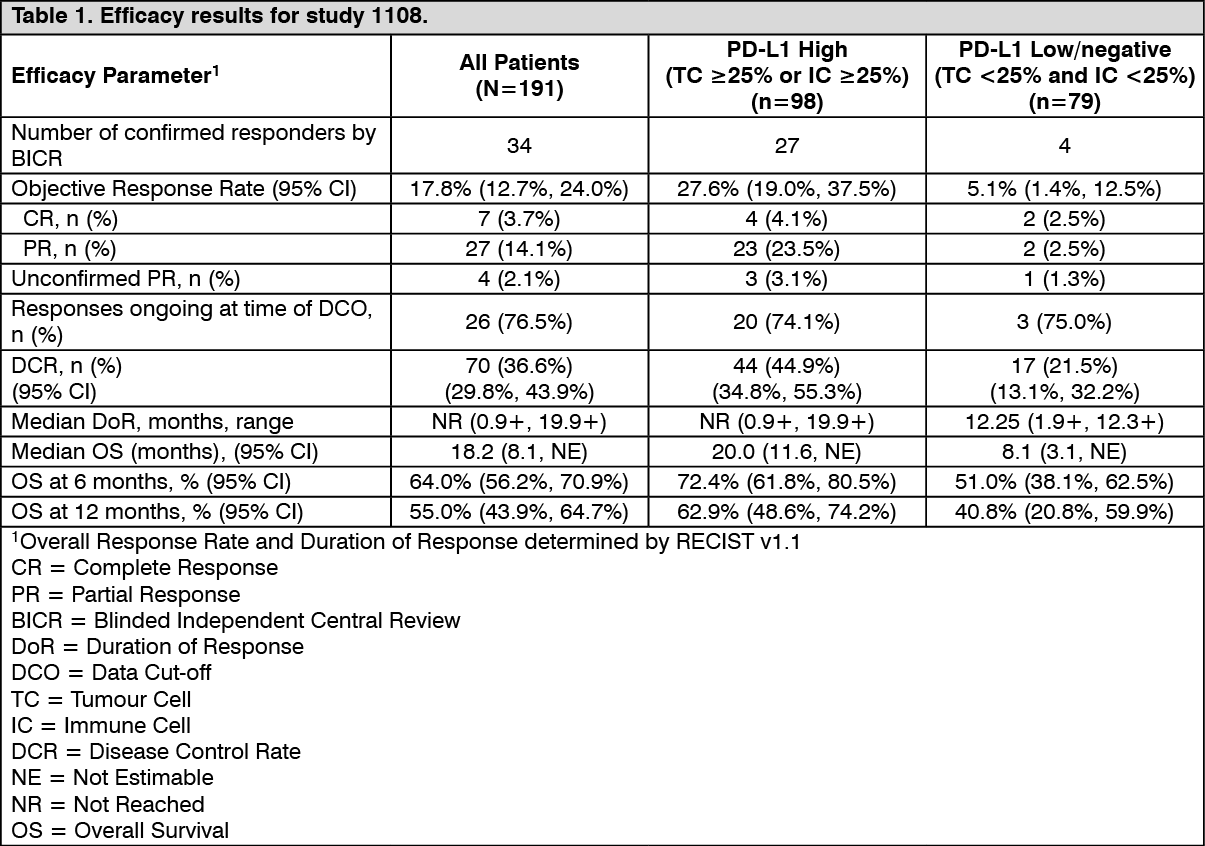
Urothelial Carcinoma - Study 1108: The efficacy of Durvalumab (IMFINZI) was evaluated in a multicentre, multi-cohort, open-label clinical trial, Study 1108. In the urothelial carcinoma cohort, 191 patients received Durvalumab (IMFINZI) 10 mg/kg every 2 weeks (Q2W) and had an opportunity to be followed for at least 16 weeks as of DCO (had tumour assessments at Weeks 6, 12, and 16). One hundred and eighty-two (182) of 191 patients with locally advanced or metastatic urothelial carcinoma had progressed while on or after a platinum-based therapy, including those patients who progressed within 12 months of receiving therapy in a neoadjuvant or adjuvant setting. Of the remaining 9 patients, 7 patients were cisplatin-ineligible at the time of study entry and 2 patients were cisplatin eligible and did not receive prior systemic therapy.
Sixty nine percent (69%) of patients received prior cisplatin, 29% had prior carboplatin and 33% received 2 or more prior lines of systemic therapy. The trial excluded patients with a history of immunodeficiency; medical conditions that required systemic immunosuppression; history of severe immune-mediated adverse reactions; untreated CNS metastases; HIV; active tuberculosis, or hepatitis B or C infection.
In this cohort, the median age was 67 years (range: 34 to 88), 71% were male, 71% were Caucasian. Ninety-three percent (93%) had visceral metastasis, including 43% with liver metastasis. Lymph-node-only metastasis was present in 7% of patients. ECOG performance status 0 (33.5%) or 1 (66.5%). Forty-three percent (43%) had a baseline creatinine clearance of <60 mL/min. The median duration of follow-up for this cohort was 5.78 months (range: 0.24 to 25.9).
All patients received Durvalumab (IMFINZI) 10 mg/kg via intravenous infusion every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed disease progression. Tumour assessments were performed at Weeks 6, 12 and 16, then every 8 weeks for the first year and every 12 weeks thereafter. The primary efficacy endpoint was Objective Response Rate (ORR) according to RECIST v1.1 as assessed by Blinded Independent Central Review (BICR). Additional efficacy endpoint included Duration of Response (DoR).
Tumour specimens were evaluated for PD-L1 expression on tumour cells (TC) and immune cells (IC) using the Ventana PD-L1 (SP263) Assay. All testing was performed prospectively at a central laboratory. Of the 191 patients, 98 were classified as PD-L1 high (TC ≥25% or IC ≥25%), 79 as PD-L1 low/negative (TC <25% and IC <25%) and samples for 14 patients were inadequate for evaluation. Table 5 summarises the efficacy results. PD-L1 high expression in patients with urothelial carcinoma was associated with numerically increased ORR. The median duration of response has not been reached.
Eight patients in the UC cohort did not have ongoing responses at the time of DCO. Seven patients progressed per BICR after an initial response, and 1 patient had PR per BICR based on the last available evaluable disease assessment (Day 168); however, the patient discontinued from treatment due to PD assessed by investigator, did not have any follow-up scans after discontinuation, and died due to disease progression on Day 608.
Among the total 34 responding patients, 76.5% (26/34) had ongoing responses at the time of analysis for ORR, 17 patients had ongoing responses of 6 months or longer, 10 patients had ongoing responses of 9 months or longer and 5 patients has ongoing responses of 12 months or longer. Of the 7 patients who progressed per BICR after an initial response, 3 patients remain on Durvalumab (IMFINZI) 10 mg/kg Q2W, and 4 patients completed 12 months of treatment with Durvalumab (IMFINZI). (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Locally Advanced NSCLC - PACIFIC Study: The efficacy of Durvalumab (IMFINZI) was evaluated in the PACIFIC Study, a randomised, double-blind, placebo-controlled, multicentre study in 713 patients with histologically or cytologically confirmed locally advanced, unresectable NSCLC. Patients had completed at least 2 cycles of definitive platinum-based chemoradiation within 1 to 42 days prior to initiation of the study and had a ECOG performance status of 0 or 1. Ninety-two percent of patients had received a total dose of 54 to 66 Gy of radiation. The study excluded patients who had progressed following chemoradiation therapy, patients with active or prior documented autoimmune disease within 2 years of initiation of the study; a history of immunodeficiency; a history of severe immune-mediated adverse reactions; medical conditions that required systemic immunosuppression, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection or patients receiving live attenuated vaccine within 30 days before or after the start of Durvalumab (IMFINZI). Patients were randomised 2:1 to receive 10 mg/kg Durvalumab (IMFINZI) (n=476) or 10 mg/kg placebo (n=237) via intravenous infusion every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed disease progression. Randomisation was stratified by gender, age (<65 years vs. ≥65 years) and smoking status (smoker vs. non- smoker). Patients with disease control at 12 months were given the option to be re-treated upon disease progression. Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter.
The demographics and baseline disease characteristics were well balanced between study arms. Baseline demographics of the overall study population were as follows: male (70%), age ≥65 years (45%), white (69%), Asian (27%), other (4%), current smoker (16%), past-smoker (75%), and never smoker (9%), WHO/ECOG PS 0 (49%), WHO/ECOG PS 1 (51%). Disease characteristics were as follows: Stage IIIA (53%), Stage IIIB (45%), histological sub-groups of squamous (46%), non-squamous (54%), PD-L1 expression TC ≥25% (22%), PD-L1 expression TC <25% (41%). (PD-L1 status was retrospectively analysed in 451 patients with available samples, taken prior to concurrent chemoradiation therapy).
The two primary endpoints of the study were overall survival (OS) and progression-free survival (PFS) of Durvalumab (IMFINZI) vs. placebo. Secondary efficacy endpoints included ORR, DoR and Time to Death or Distant Metastasis (TTDM). PFS, ORR, DoR and TTDM were assessed by BICR according to RECIST v1.1.
The study demonstrated a statistically significant and clinically meaningful improvement in OS in the Durvalumab (IMFINZI)-treated group compared with the placebo group [HR=0.68 (95% CI: 0.53, 0.87), p=0.00251]. Median OS was not reached in the Durvalumab (IMFINZI)-treated group and was 28.7 months in the placebo group. The study demonstrated a statistically significant and clinically meaningful improvement in PFS in the Durvalumab (IMFINZI)-treated group compared with the placebo group [hazard ratio (HR)=0.52 (95% CI: 0.42, 0.65), p <0.0001]. Median PFS was 16.8 months in the Durvalumab (IMFINZI)-treated group and 5.6 months in the placebo group. See Table 2 and Figures 2 and 3.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The improvements in OS and PFS in favour of patients receiving Durvalumab (IMFINZI) compared to those receiving placebo were consistently observed across predefined subgroups analysed. Sensitivity analyses of OS and PFS demonstrated a consistent treatment effect with that observed in the primary analysis.
Patient-reported outcomes: Patient-reported symptoms, function and HRQoL were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 and C30 were assessed at baseline, every 4 weeks for the first 8 weeks, followed by every 8 weeks until completion of the treatment period or discontinuation of study drug due to toxicity or disease progression. Compliance was high and very similar between the Durvalumab (IMFINZI) and placebo treatment groups.
At baseline, no differences in patient-reported symptoms, function and HRQoL were observed between Durvalumab (IMFINZI) and placebo groups. Throughout the duration of the study to Week 48, there was no clinically meaningful difference between Durvalumab (IMFINZI) and placebo groups in symptoms, functioning and HRQoL (as assessed by a difference of greater than or equal to 10 points).
Metastatic NSCLC - POSEIDON Study: POSEIDON was a study designed to evaluate the efficacy of Durvalumab (IMFINZI) with or without tremelimumab in combination with platinum-based chemotherapy. POSEIDON was a randomised, open-label, multicentre study in 1013 metastatic NSCLC patients with no sensitizing epidermal growth factor receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) genomic tumour aberrations. Patients with a histologically or cytologically documented metastatic NSCLC were eligible for enrolment. Patients had no prior chemotherapy or any other systemic therapy for metastatic NSCLC. Prior to randomisation, patients had tumour PD-L1 status confirmed by using the Ventana PD-L1 (SP263) Assay. Patients had a World Health Organization/Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1.
The study excluded patients with active or prior documented autoimmune disease; active and/or untreated brain metastases; a history of immunodeficiency; administration of systemic immunosuppression within 14 days before the start of Durvalumab (IMFINZI) or tremelimumab, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of Durvalumab (IMFINZI) and/or tremelimumab.
Randomisation was stratified by tumour cells (TC) PD-L1 expression (TC≥50% vs. TC<50%), disease stage (Stage IVA vs. Stage IVB), and histology (non-squamous vs. squamous).
Patients were randomised 1:1:1 to receive: Arm 1: Durvalumab (IMFINZI) 1500 mg with tremelimumab 75 mg and platinum-based chemotherapy every 3 weeks for 4 cycles, followed by, Durvalumab (IMFINZI) 1500 mg every 4 weeks as monotherapy. A fifth dose of tremelimumab 75 mg was given at Week 16 alongside Durvalumab (IMFINZI) dose 6.
Arm 2: Durvalumab (IMFINZI) 1500 mg and platinum-based chemotherapy every 3 weeks for 4 cycles, followed by, Durvalumab (IMFINZI) 1500 mg every 4 weeks as monotherapy.
Arm 3: Platinum-based chemotherapy every 3 weeks for 4 cycles as monotherapy. Patients could receive additional 2 cycles (a total of 6 cycles post-randomisation), as clinically indicated, at Investigator's discretion.
In the 3 treatment arms, patients received one of the following histology-based chemotherapy regimens: Non-squamous NSCLC: Pemetrexed 500 mg/m
2 with carboplatin AUC 5-6 or cisplatin 75 mg/m
2 every 3 weeks, unless contraindicated by the investigator, pemetrexed maintenance could be given.
Squamous NSCLC: Gemcitabine 1000 or 1250 mg/m
2 on Days 1 and 8 with cisplatin 75 mg/m
2 or carboplatin AUC 5-6 on Day 1 every 3 weeks.
Non-squamous and Squamous NSCLC: Nab-paclitaxel 100 mg/m
2 on Days 1, 8, and 15 with carboplatin AUC 5-6 on Day 1 every 3 weeks.
Tremelimumab was given up to a maximum of 5 doses unless there was disease progression or unacceptable toxicity. Durvalumab (IMFINZI) and histology-based pemetrexed maintenance therapy (when applicable) was continued until disease progression or unacceptable toxicity. Administration of Durvalumab (IMFINZI) monotherapy was permitted beyond disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator. Patients with disease progression during Durvalumab (IMFINZI) monotherapy were given the option to be retreated with 4 additional cycles of tremelimumab alongside Durvalumab (IMFINZI).
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The dual primary endpoints of the study were PFS and Overall Survival OS for Durvalumab (IMFINZI) + platinum-based chemotherapy (Arm 2) vs. platinum-based chemotherapy alone (Arm 3). The key secondary endpoints of the study were PFS and OS for Durvalumab (IMFINZI) + tremelimumab + platinum-based chemotherapy (Arm 1) vs. platinum-based chemotherapy alone (Arm 3). The secondary endpoints included ORR and DoR. PFS, ORR, and DoR were assessed using BICR according to RECIST v1.1. At planned analyses for OS and PFS, Durvalumab (IMFINZI) + tremelimumab + platinum-based chemotherapy (Arm 1) vs. platinum-based chemotherapy (Arm 3) met the efficacy boundaries for the endpoints of OS and PFS. The results are summarised as follows.
The demographics and baseline disease characteristics were generally well-balanced between study arms. Baseline demographics of the overall study population were as follows: male (76.0%), age ≥65 years (47.1%), white (55.9%), Asian (34.6%), black or African American (2.0%), other (7.6%), non-Hispanic or Latino (84.2%), current smoker or past-smoker (78.0%), and never smoker (21.9%), WHO/ECOG PS 0 (33.4%), WHO/ECOG PS 1 (66.5%). Disease characteristics were as follows: Stage IVA (50.0%), Stage IVB (49.6%), histological sub-groups of squamous (36.9%), non-squamous (62.9%), PD-L1 expression TC≥50% (28.8%), PD-L1 expression TC <50% (71.1%).
The study demonstrated a statistically significant and clinically meaningful improvement in OS in the Durvalumab (IMFINZI) + tremelimumab + platinum-based chemotherapy (Arm 1) vs. platinum-based chemotherapy alone (Arm 3) [HR=0.77 (95% CI: 0.650, 0.916), p=0.00304]. Durvalumab (IMFINZI) + tremelimumab + platinum-based chemotherapy demonstrated a statistically significant and clinically meaningful improvement in PFS vs. platinum-based chemotherapy alone (Arm 3) [HR=0.72 (95% CI: 0.600, 0.860), p=0.00031]. See Table 3 and Figures 4 and 5.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Subgroup analysis: The improvements in OS and PFS favour patients receiving Durvalumab (IMFINZI) + tremelimumab + platinum-based chemotherapy compared to those receiving platinum-based chemotherapy alone and were consistently observed across the prespecified subgroups based on demographic and baseline characteristics, biomarker status, histology, planned chemotherapy, and disease characteristics. An exception was noted in the never smoker subgroup for OS. However, due to the small numbers of patients, no definitive conclusions can be drawn from these data.
Patient Reported Outcomes: Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). Both questionnaires were administered up to second disease progression (PFS2) or death (whichever came first). At baseline, patient-reported symptoms, functioning or HRQoL scores were comparable between the study arms. Overall compliance for EORTC QLQ-C30 and EORTC QLQ-L13 were 73.0% and 72.8% in the T + D + SoC arm and 65.0% and 64.8% in the SoC chemotherapy arm.
Delay in time to deterioration (TTD) of symptoms, functioning, and global health status/QoL: D+T+SoC prolonged the median TTD in patient-reported symptoms, functioning and global health status/QoL compared to SoC alone (see Tables 4 and 5). Nominally significant differences in TTD in favour of T + D + SoC compared to SoC alone were observed for the pre-specified domains of interest of global health status/QoL, physical functioning and dyspnea (EORTC QLQ-LC13) (HRs ranging from 0.75 to 0.78; nominal p-values <0.05). (See Tables 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
SCLC - CASPIAN Study: CASPIAN was a study designed to evaluate the efficacy of Durvalumab (IMFINZI) with or without tremelimumab in combination with etoposide and either carboplatin or cisplatin. CASPIAN was a randomised, open-label, multicentre study in 805 treatment naïve ES-SCLC patients with WHO/ECOG Performance status of 0 or 1, suitable to receive a platinum-based chemotherapy regimen as first-line treatment for SCLC, with life expectancy ≥12 weeks, at least one target lesion by RECIST 1.1 and adequate organ and bone marrow function. Patients with asymptomatic or treated brain metastases were eligible. The study excluded patients with a history of chest radiation therapy; a history of active primary immunodeficiency; autoimmune disorders including paraneoplastic syndrome (PNS); active or prior documented autoimmune or inflammatory disorders; use of systemic immunosuppressants within 14 days before the first dose of the treatment except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of Durvalumab (IMFINZI).
Randomisation was stratified by the planned platinum-based therapy in cycle 1 (carboplatin or cisplatin).
Patients were randomised 1:1:1 to receive: Arm 1: Durvalumab (IMFINZI) 1500 mg + tremelimumab 75 mg + etoposide and either carboplatin or cisplatin.
Arm 2: Durvalumab (IMFINZI) 1500 mg + etoposide and either carboplatin or cisplatin.
Arm 3: Either carboplatin (AUC 5 or 6 mg/mL/min) or cisplatin (75-80 mg/m
2) on Day 1 and etoposide (80-100 mg/m
2) intravenously on Days 1, 2, and 3 of each 21-day cycle for between 4-6 cycles.
For patients randomised to Arm 1 and 2, etoposide and either carboplatin or cisplatin was limited to 4 cycles on an every 3 week schedule subsequent to randomisation. Durvalumab (IMFINZI) monotherapy continued until disease progression or unacceptable toxicity. Administration of Durvalumab (IMFINZI) monotherapy was permitted beyond disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator.
Patients randomised to Arm 3, were permitted to receive a total of up to 6 cycles of etoposide and either carboplatin or cisplatin. After completion of chemotherapy, prophylactic cranial irradiation (PCI) was permitted only in Arm 3 per investigator discretion.
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The primary endpoints of the study were OS of Durvalumab (IMFINZI) + chemotherapy (Arm 2) vs. chemotherapy alone (Arm 3) and Durvalumab (IMFINZI) + tremelimumab + chemotherapy (Arm 1) vs. chemotherapy alone (Arm 3). The key secondary endpoint was PFS. Other secondary endpoints were ORR, OS and PFS landmarks and Patient-Reported Outcomes (PRO). PFS and ORR were assessed using Investigator assessments according to RECIST v1.1.
At a planned interim analysis, Durvalumab (IMFINZI) + chemotherapy (Arm 2) vs chemotherapy (Arm 3) met the efficacy boundary of the primary endpoint of OS. The results are summarised as follows.
The demographics and baseline disease characteristics were well balanced between the two study arms (268 patients in Arm 2 and 269 patients in Arm 3). Baseline demographics of the overall study population were as follows: male (69.6%), age ≥65 years (39.6%), median age 63 years (range: 28 to 82 years), white (83.8%), Asian (14.5%), black or African American (0.9%), other (0.6 %), non-Hispanic or Latino (96.1%), current or past-smoker (93.1%), never smoker (6.9%), WHO/ECOG PS 0 (35.2%), WHO/ECOG PS 1 (64.8%), Stage IV 90.3%, 24.6% of the patients received cisplatin and 74.1% of the patients received carboplatin. In Arm 3, 56.8% of the patients received 6 cycles of chemotherapy and 7.8% of the patients received PCI.
The study demonstrated a statistically significant and clinically meaningful improvement in OS with Durvalumab (IMFINZI) + chemotherapy (Arm 2) vs. chemotherapy alone (Arm 3) [HR=0.73 (95% CI: 0.591, 0.909), p=0.0047]. Durvalumab (IMFINZI) + chemotherapy demonstrated an improvement in PFS vs. chemotherapy alone [HR=0.78 (95% CI: 0.645, 0.936) nominal p-value=0.0078]. See Table 6 and Figures 6 and 7.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Subgroup analysis: The improvements in OS in favour of patients receiving Durvalumab (IMFINZI) + chemotherapy compared to those receiving chemotherapy alone, were consistently observed across the prespecified subgroups based on demographics, geographical region, carboplatin or cisplatin use and disease characteristics.
Patient-Reported Outcomes: Patient-reported symptoms, function and HRQoL were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). Both questionnaires were administered up to second disease progression (PFS2) or death (whichever came first). At baseline, patient-reported symptoms, functioning or HRQoL scores were comparable between the study arms. Compliance was 60% or higher over 84 weeks in Durvalumab (IMFINZI) + chemotherapy and 20 weeks in the chemotherapy only arm.
Delay in time to deterioration of symptoms, functioning, and global health status/QoL: Durvalumab (IMFINZI) + chemotherapy demonstrated improvement by delaying time to deterioration in a broad range of patient-reported symptoms, function, and global health status/QoL compared to chemotherapy alone (see Tables 7 and 8).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Change from baseline in lung cancer symptoms over 12 months (mixed model for repeated measures): Durvalumab (IMFINZI) + chemotherapy improved appetite loss by demonstrating a statistically significant difference in mean change from baseline versus chemotherapy alone during the overall time period from randomisation until 12 months (Estimated mean difference -4.5; 99% CI -9.04, -0.04; p=0.009). Both treatment arms demonstrated numerical symptom reduction in cough, chest pain, dyspnoea and fatigue over the same time period.
Patient-reported outcome results should be interpreted in the context of the open-label study design.
BTC - TOPAZ-1 Study: TOPAZ-1 was a study designed to evaluate the efficacy of Durvalumab (IMFINZI) in combination with gemcitabine and cisplatin. TOPAZ-1 was a randomised, double-blind, placebo-controlled, multicentre study in 685 patients with histologically confirmed locally advanced or metastatic BTC and ECOG performance status of 0 or 1. Patients who developed recurrent disease more than 6 months after surgery and/or completion of adjuvant therapy were included. Patients must have had at least one target lesion by RECIST v1.1 and adequate organ and bone marrow function.
The study excluded patients with ampullary carcinoma, active or prior documented autoimmune or inflammatory disorders, HIV infection or active infections, including tuberculosis or hepatitis C or patients with current or prior use of immunosuppressive medication within 14 days before the first dose of Durvalumab (IMFINZI).
Randomisation was stratified by disease status and primary tumour location.
Patients were randomised 1:1 to receive: Arm 1: Durvalumab (IMFINZI) 1500 mg administered intravenously on Day 1+ gemcitabine 1000 mg/m
2 and cisplatin 25 mg/m
2 (each administered on Days 1 and 8) every 3 weeks (21 days) for up to 8 cycles, followed by Durvalumab (IMFINZI) 1500 mg every 4 weeks as long as clinical benefit is observed or until unacceptable toxicity, or; Arm 2: Placebo administered intravenously on Day 1+ gemcitabine 1000 mg/m
2 and cisplatin 25 mg/m
2 (each administered on Days 1 and 8) every 3 weeks (21 days) for up to 8 cycles, followed by placebo every 4 weeks as long as clinical benefit is observed or until unacceptable toxicity.
Tumour assessments were conducted every 6 weeks for the first 24 weeks after the date of randomisation, and then every 8 weeks until confirmed objective disease progression.
The primary endpoint of the study was OS and the key secondary endpoint was PFS. Other secondary endpoints were ORR, DoR and PRO. PFS, ORR and DoR were Investigator assessed according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced between the two study arms (341 patients in Arm 1 and 344 patients in Arm 2). Baseline demographics of the overall study population were as follows: male (50.4%), age <65 years (53.3%), white (37.2%), Asian (56.4%), black or African American (2.0%), other (4.2%), non-Hispanic or Latino (93.1%), ECOG PS 0 (49.1%), vs. PS 1 (50.9%), primary tumour location intrahepatic cholangiocarcinoma (55.9%), extrahepatic cholangiocarcinoma (19.1%) and gallbladder cancer (25.0%), disease status recurrent (19.1%) vs. initially unresectable (80.7%), metastatic (86.0%) vs locally advanced (13.9%).
The study demonstrated a statistically significant and clinically meaningful improvement in OS and PFS at a pre-planned interim analysis based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary and the actual number of events observed (Lan◦and◦DeMets 1983). The results in OS were [HR=0.80, (95% CI: 0.66, 0.97), p=0.021] and in PFS [HR=0.75, (95% CI: 0.63, 0.89), p=0.001]. The maturity for OS was 61.9% and the maturity for PFS was 83.6%. The boundary for declaring statistical significance for OS was 0.03 for an 4.9% overall alpha. Results from this analysis for PFS, ORR and DoR are presented in Table 9. PFS is also presented in Figure 9.
An additional OS analysis was performed 6.5 months after the interim analysis with an OS maturity of 76.9%. The observed treatment effect was consistent with the interim analysis. The OS HR was 0.76 (95% CI: 0.64, 0.91) and median survival was 12.9 months (95% CI: 11.6, 14.1). Results from this analysis for OS are presented in the Table 9 and Figure 8. (See Table 9 and Figures 8 and 9.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Subgroup analysis: The improvements in OS and PFS in favour of patients receiving Durvalumab (IMFINZI) + chemotherapy compared to those receiving placebo + chemotherapy, were consistently observed across the prespecified subgroups based on demographics, geographical region, primary tumour location, disease status, ECOG PS, and PD-L1 expression levels.
Patient-Reported Outcomes: Patient-reported symptoms, function and global health status/QoL (GHS/QoL) were collected using the EORTC QLQ-C30 and its biliary tract cancer module (EORTC QLQ-BIL21). At baseline, patient-reported symptoms, functioning and GHS/QoL scores were comparable between the study arms. Time to deterioration and change from baseline analyses were consistent with no detriment in symptoms, function and GHS/QoL per EORTC QLQ-C30 and EORTC QLQ-BIL21 in the Durvalumab (IMFINZI) + chemotherapy group compared to the placebo + chemotherapy group.
HCC - HIMALAYA Study: The efficacy of STRIDE was evaluated in the HIMALAYA study, a randomised, open-label, multicentre study in patients with confirmed uHCC who did not receive prior systemic treatment for HCC. The study included patients with BCLC Stage C or B (not eligible for locoregional therapy) and Child-Pugh Score Class A.
The study excluded patients with co-infection of viral hepatitis B and hepatitis C; active or prior documented GI bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Patients with esophageal varices were included except those with active or prior documented GI bleeding within 12 months prior to study entry.
Randomisation was stratified by macrovascular invasion (MVI) (yes vs. no), etiology of liver disease (confirmed hepatitis B virus vs. confirmed hepatitis C virus vs. others) and ECOG performance status (0 vs. 1).
The HIMALAYA study randomised 1171 patients 1:1:1 to receive: Durvalumab (IMFINZI): durvalumab 1500 mg every 4 weeks; STRIDE: tremelimumab 300 mg as a single priming dose + Durvalumab (IMFINZI) 1500 mg; followed by Durvalumab (IMFINZI) 1500 mg every 4 weeks; S: Sorafenib 400 mg twice daily.
Treatment continued as long as clinical benefit was observed or until unacceptable toxicity. Patients in all arms could continue to receive treatment after evidence of disease progression if, in the Investigator's opinion, they were benefiting from study drug and met all inclusion and exclusion criteria for treatment beyond progression. In addition, patients in the STRIDE arm who continued treatment beyond progression were allowed to be rechallenged once with an additional single dose of tremelimumab 300 mg after cycle five of Durvalumab (IMFINZI). Of the 182 patients enrolled to the STRIDE arm who received Durvalumab (IMFINZI) beyond progression, the median OS was 19.5 months (95% CI: 15.4, 23.4). Of the 30 patients who were enrolled to the STRIDE arm who were rechallenged with tremelimumab, the median OS was 30.4 months (95% CI: 23.4, NR).
Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter. Survival assessments were conducted every month for the first 3 months following treatment discontinuation and then every 2 months.
The primary endpoint was OS. Key secondary endpoints were PFS, Investigator assessed ORR and DoR according to RECIST v1.1. PROs were also assessed.
The demographics and baseline disease characteristics were generally representative for patients with uHCC. The baseline demographics of the overall study population were as follows: male (83.7%), age <65 years (50.4%), white (44.6%), Asian (50.7%), black or African American (1.7%), other (2.3%), ECOG PS 0 (62.6%); Child-Pugh Class score A (99.5%), macrovascular invasion (25.2%), extrahepatic spread (53.4%), viral etiology; hepatitis B (30.6%), hepatitis C (27.2%), uninfected (42.2%).
The study demonstrated a statistically significant and clinically meaningful improvement in OS with STRIDE vs. S [HR=0.78 [95% CI 0.66, 0.92]; p=0.0035]. See Table 10 and Figure 10.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patient reported outcomes: Patient-reported symptoms, function and HRQoL were collected using the EORTC QLQ-C30 and its hepatocellular carcinoma module (EORTC QLQ-HCC18). At baseline, patient-reported symptoms, functioning or HRQoL scores were comparable between the study arms.
Delay in time to deterioration of symptoms, functioning, and global health status/QoL: STRIDE vs. S demonstrated a clinically meaningful improvement by delaying time to deterioration in a broad range of patient-reported symptoms, function, and global health status/QoL compared to S. Longer time to deterioration (median in months) was observed in the STRIDE arm compared to S for the following symptoms: Global Health Status (7.5 vs. 5.7 months, HR 0.76, p=0.0306); physical functioning (12.9 vs. 7.4 months, HR 0.68; p=0.0020), fatigue (7.4 vs. 5.4 months, HR 0.71; p=0.0026), nausea (25.0 vs. 11.0 months, HR 0.65; p=0.0033), appetite loss (12.6 vs. 6.9 months, HR 0.59; p <0.0001), abdominal pain (16.8 vs. 8.9 months, HR 0.61; p=0.0008) and abdominal swelling (20.9 vs. 11.1 months, HR 0.74; p=0.0431.
Change from baseline in patient-reported symptoms (mixed model for repeated measures): STRIDE improved patient-reported HRQoL functioning and diarrhoea by demonstrating a nominal difference and clinically meaningful mean change from baseline vs. S from randomisation until 8 months (Estimated mean difference at 8 months: -18.5 95% CI: -23.24, -13.84 and p-value: <0.0001).
Patient-reported outcome results should be interpreted in the context of the open-label study design.
HCC - Study 22: The safety and efficacy of STRIDE was evaluated in Study 22, an open-label, multi-part, multicentre study in 75 immunotherapy naïve patients with uHCC who had progressed on, are intolerant to, or have refused sorafenib. The study included patients with BCLC Stage C or B (not eligible for locoregional therapy), ECOG performance status of 0 or 1 and Child-Pugh Score Class A.
The study excluded patients with co-infection of viral hepatitis B and hepatitis C; active or prior documented GI bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Treatment continued as long as clinical benefit was observed or until unacceptable toxicity. Patients who completed the assigned dosing cycles and were benefiting from study drug in the Investigator's opinion and subsequently had evidence of disease progression during the Durvalumab (IMFINZI) monotherapy phase could be rechallenged with tremelimumab 300 mg.
Tumour assessments were conducted every 8 weeks.
The primary objective was safety and tolerability. Key secondary endpoints included OS, ORR and DoR. ORR, DoR and PFS were based on Investigator assessments and BICR according to RECIST 1.1.
The baseline demographics of the study population (STRIDE) were as follows: male (86.7%); age <65 years (45.3%), white (36.0%); Asian (58.7%); black or African American (5.3%); other (0%), ECOG PS 0 (61.3%), Child-Pugh Class/Score A/5 (68.0%), Child-Pugh Class/Score A/6 (30.7%), macrovascular invasion (21.3%); extrahepatic spread (70.7%), viral etiology; hepatitis B (36.0%), hepatitis C (28.0%), uninfected (36.0%); prior systemic therapy (73.3%).
Efficacy results are shown in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics (PK) of durvalumab was assessed for Durvalumab (IMFINZI) as a single agent, in combination with chemotherapy, in combination with tremelimumab and platinum-based chemotherapy, and in combination with tremelimumab.
The pharmacokinetics of durvalumab was studied in patients with solid tumours with doses ranging from 0.1 to 20 mg/kg administered once every two, three or four weeks. PK exposure increased more than dose-proportionally (non-linear PK) at doses <3 mg/kg and dose proportionally (linear PK) at doses ≥3 mg/kg. Steady state was achieved at approximately 16 weeks. Based on population PK analysis that included patients in the dose range of 10 mg/kg Q2W, 15 mg/kg Q3W and 20 mg/kg Q4W, the geometric mean, steady state volume of distribution (Vss) was 5.64 L. Durvalumab clearance (CL) decreased over time resulting in a geometric mean steady state clearance (CLss) of 8.16 mL/h at Day 365; the decrease in CLss was not considered clinically relevant. The terminal half-life (t1/2), based on baseline CL, was approximately 18 days. There was no clinically meaningful difference between the PK of durvalumab as a single agent, in combination with chemotherapy, in combination with tremelimumab and platinum-based chemotherapy or in combination with tremelimumab.
Special populations: Age (19-96 years), body weight (31-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, soluble PD-L1, tumour type, race, mild renal impairment (creatinine clearance (CRCL) 60 to 89 mL/min), moderate renal impairment (creatinine clearance (CRCL) 30 to 59 mL/min), mild hepatic impairment (bilirubin ≤ ULN and AST >ULN or bilirubin >1.0 to 1.5 x ULN and any AST), moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST) or ECOG/WHO status had no clinically significant effect on the pharmacokinetics of durvalumab.
The effect of severe renal impairment (CRCL 15 to 29 mL/min) or severe hepatic impairment (bilirubin >3.0 x ULN and any AST) on the pharmacokinetics of durvalumab is unknown.
Elderly: No dose adjustment is required for elderly patients (≥65 years of age). Of the 191 patients with urothelial carcinoma (primary efficacy population) treated with Durvalumab (IMFINZI), 118 patients were 65 years or older. No overall clinically meaningful differences in safety or efficacy were reported between patients ≥65 years of age and younger patients.
Of the 476 patients with locally advanced, unresectable NSCLC (primary efficacy population) treated with Durvalumab (IMFINZI), 215 patients were 65 years or older. No overall clinically meaningful differences in safety were reported between patients ≥65 years of age and younger patients.
Of the 265 patients with ES-SCLC treated with Durvalumab (IMFINZI) in combination with chemotherapy, 101 (38%) patients were 65 years or older. There were no overall clinically meaningful differences in safety or effectiveness between patients ≥65 years of age and younger patients.
Of the 338 patients with metastatic NSCLC treated with Durvalumab (IMFINZI) in combination with tremelimumab and platinum-based chemotherapy, 147 (43%) patients were 65 years or older. There were no overall clinically meaningful differences in safety or effectiveness between patients ≥65 years of age and younger patients.
Of the 462 patients with uHCC treated with STRIDE, 173 (37.4%) patients were 65 years or older and 63 (13.6%) patients were 75 years or older. There were no clinically meaningful differences in safety or efficacy between patients 65 years or older and younger patients.
Of the 338 patients with BTC treated with Durvalumab (IMFINZI) in combination with chemotherapy, 158 (46.7%) patients were 65 years or older. There were no overall clinically meaningful differences in safety or effectiveness between patients ≥65 years of age and younger patients.
Drug interaction studies: PK drug-drug interaction between durvalumab and chemotherapy was assessed in the CASPIAN study and no clinically meaningful PK drug-drug interaction was identified.
PK drug-drug interaction between durvalumab and tremelimumab and platinum-based chemotherapy was assessed in the POSEIDON study and no clinically meaningful PK drug-drug interaction was identified.
Immunogenicity: As with all therapeutic proteins, there is a potential for immunogenicity. Immunogenicity of Durvalumab (IMFINZI) as monotherapy is based on pooled data in 2280 patients who were treated with Durvalumab (IMFINZI) 10 mg/kg every 2 weeks or 20 mg/kg every 4 weeks as a single-agent and evaluable for the presence of anti-drug antibodies (ADA). Sixty-nine patients (3.0%) tested positive for treatment emergent ADA. Neutralizing antibodies against durvalumab were detected in 0.5% (12/2280) patients. The presence of ADAs did not have a clinically relevant effect on pharmacokinetics, pharmacodynamics or safety.
In the CASPIAN study, of the 201 patients who were treated with Durvalumab (IMFINZI) 1500 mg every 3 weeks in combination with chemotherapy and evaluable for the presence of ADAs, 0 (0%) patients tested positive for treatment-emergent ADAs. The impact of treatment-emergent ADA on pharmacokinetics and clinical safety of durvalumab was not evaluable as no patient samples tested positive for treatment-emergent durvalumab ADA.
In the TOPAZ-1 study, of the 240 patients who were treated with Durvalumab (IMFINZI) 1500 mg every 3 weeks in combination with chemotherapy, followed by Durvalumab (IMFINZI) 1500 mg every 4 weeks and evaluable for the presence of ADAs, 2 (0.8%) patients tested positive for treatment-emergent ADAs. There were insufficient numbers of patients with treatment emergent ADAs or neutralizing antibodies (2 patients each) to determine whether ADAs have an impact on pharmacokinetics and clinical safety of durvalumab.
In the POSEIDON study, of the 286 patients who were treated with Durvalumab (IMFINZI) 1500 mg in combination with tremelimumab every 3 weeks and platinum-based chemotherapy and evaluable for the presence of ADAs, 29 (10.1%) patients tested positive for treatment-emergent ADAs. Neutralizing antibodies against durvalumab were detected in 1% (3/286) patients. The presence of ADAs did not have an apparent effect on pharmacokinetics or safety.
In the HIMALAYA study, of the 294 patients who were treated with STRIDE and evaluable for the presence of ADAs, 9 (3.1%) patients tested positive for treatment-emergent ADAs. Neutralizing antibodies against durvalumab were detected in 1.7% (5/294) patients. The presence of ADAs did not have an apparent effect on pharmacokinetics or safety.
Immunogenicity assay results are highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease.
For these reasons, comparison of incidence of antibodies to Durvalumab (IMFINZI) with the incidence of antibodies to other products may be misleading.
Toxicology: Preclinical safety data: Carcinogenicity and mutagenicity: The carcinogenic and genotoxic potential of durvalumab has not been evaluated.
Reproductive toxicology: As reported in the literature, the PD-1/PD-L1 pathway plays a central role in preserving pregnancy by maintaining maternal immune tolerance to the foetus, and in mouse allogeneic pregnancy models disruption of PD-L1 signaling was shown to result in an increase in foetal loss. In reproduction studies in cynomolgus monkeys, administration of durvalumab from the confirmation of pregnancy through delivery at exposure levels approximately 22 times higher than those observed at the clinical dose of 10 mg/kg of durvalumab (based on AUC) was not associated with maternal toxicity or effects on embryofoetal development, pregnancy outcome or postnatal development.
Animal toxicology and/or pharmacology: Repeat dose toxicity studies in sexually mature cynomolgus monkeys with durvalumab of up to 3 months duration were not associated with any adverse effects that were considered of relevance to humans.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image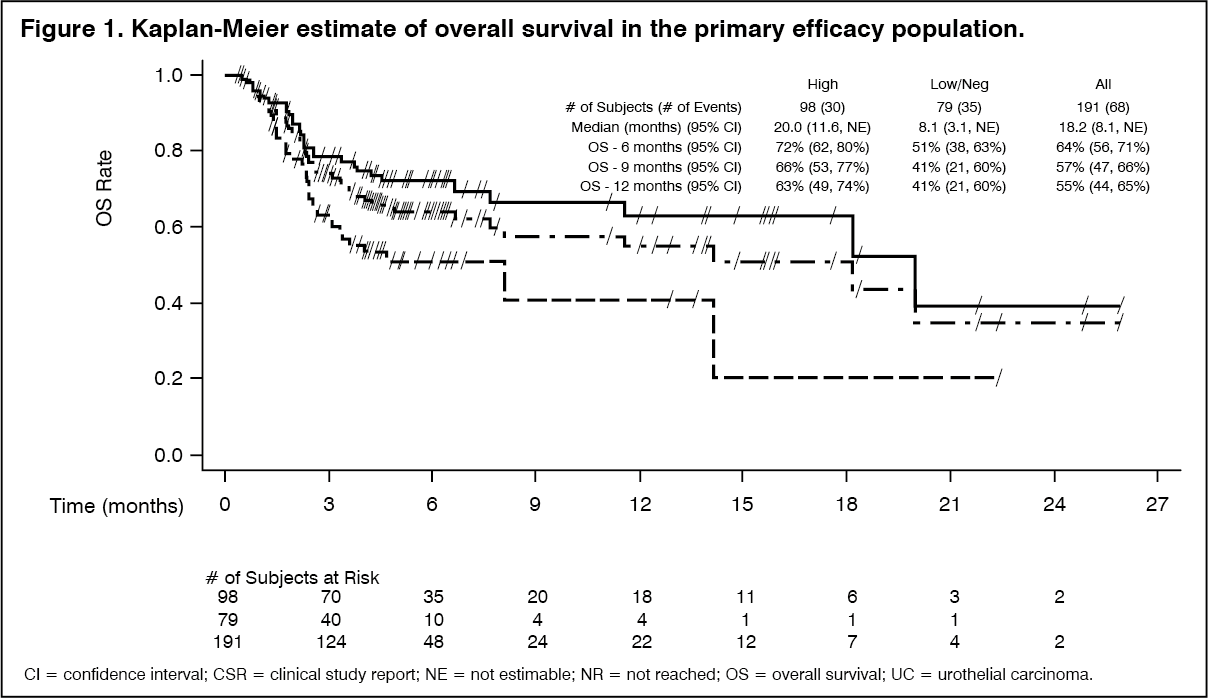 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image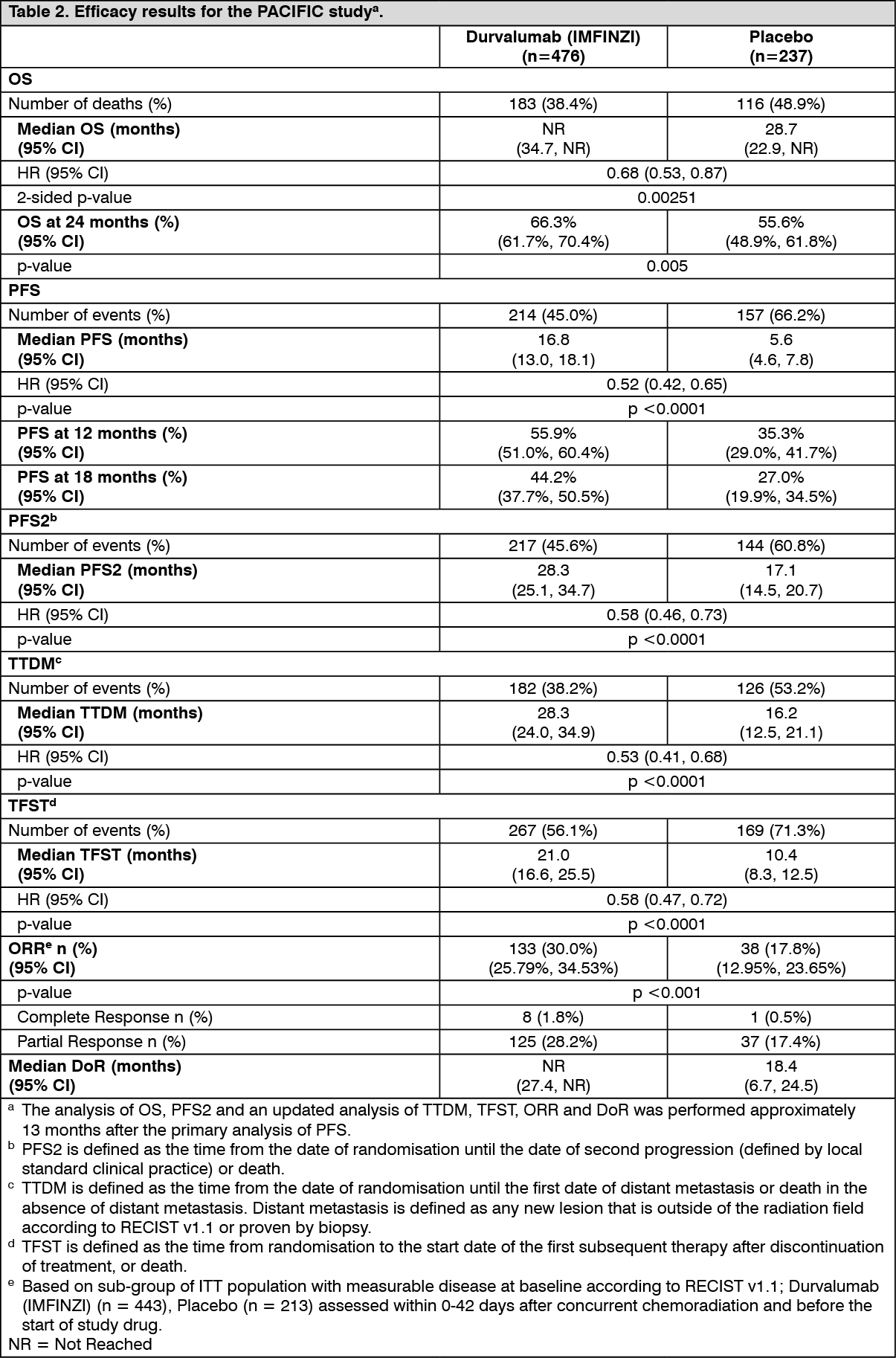 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image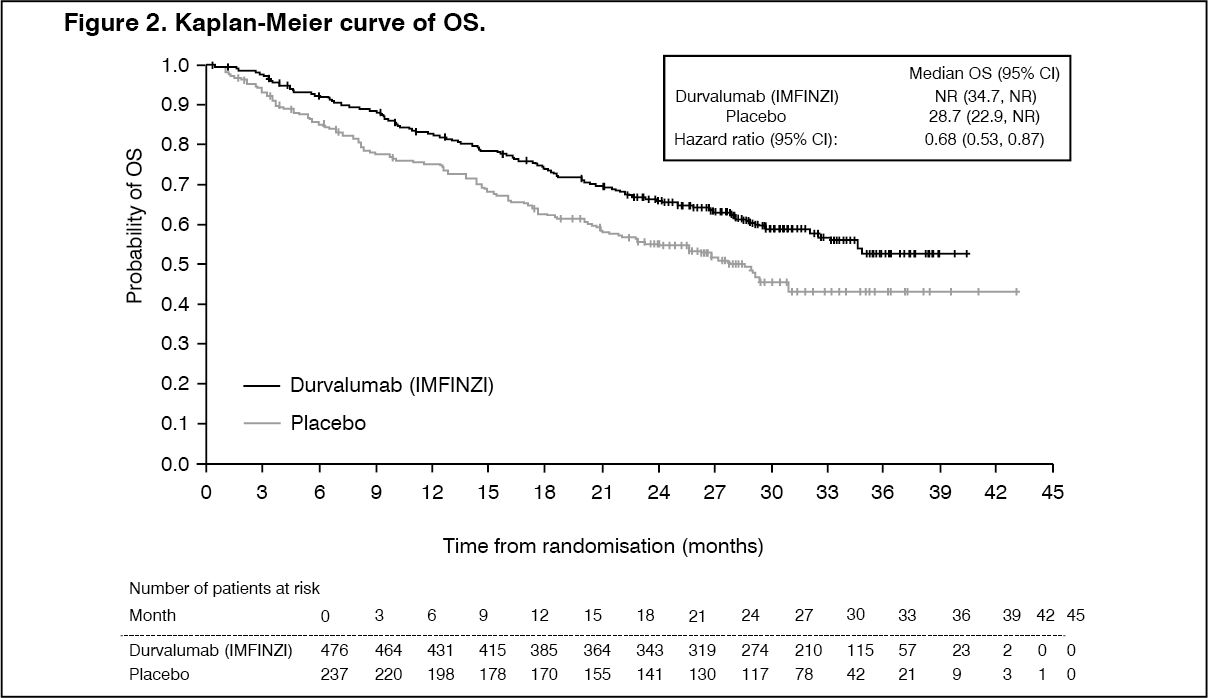 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image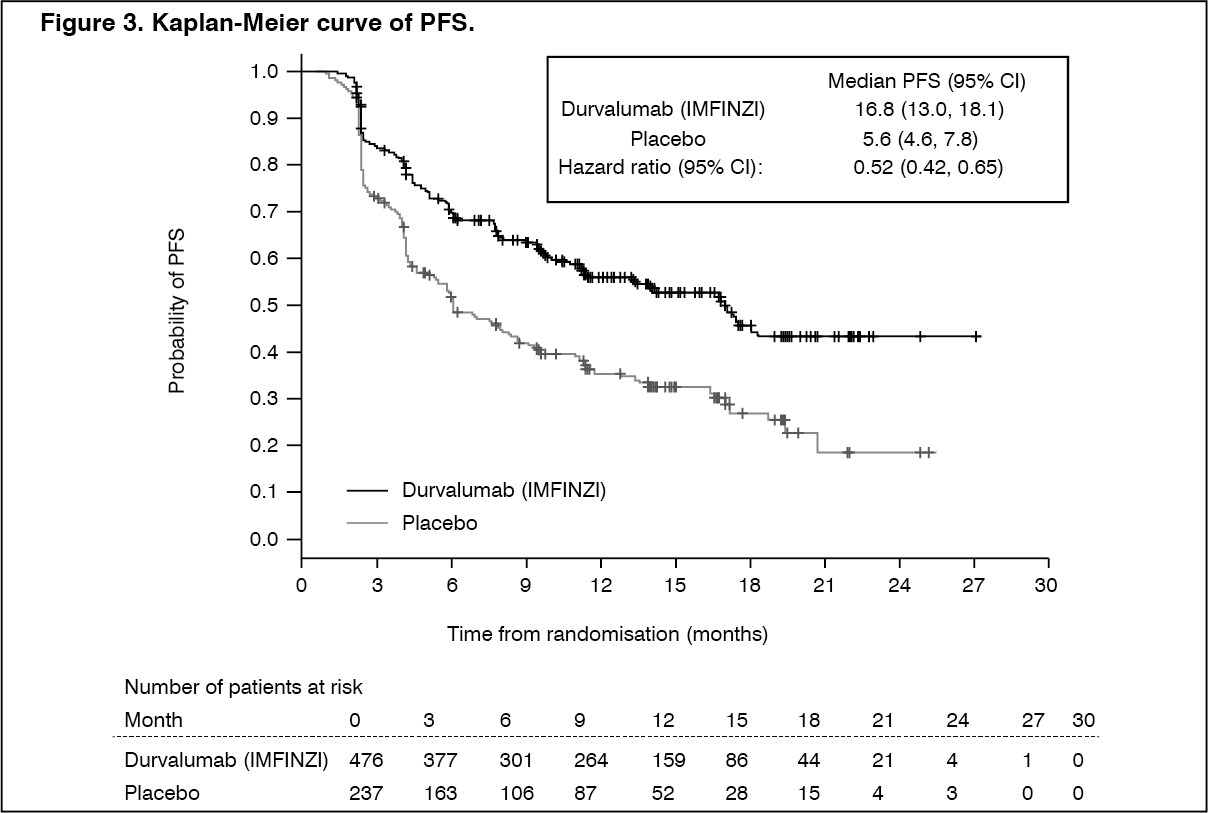 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image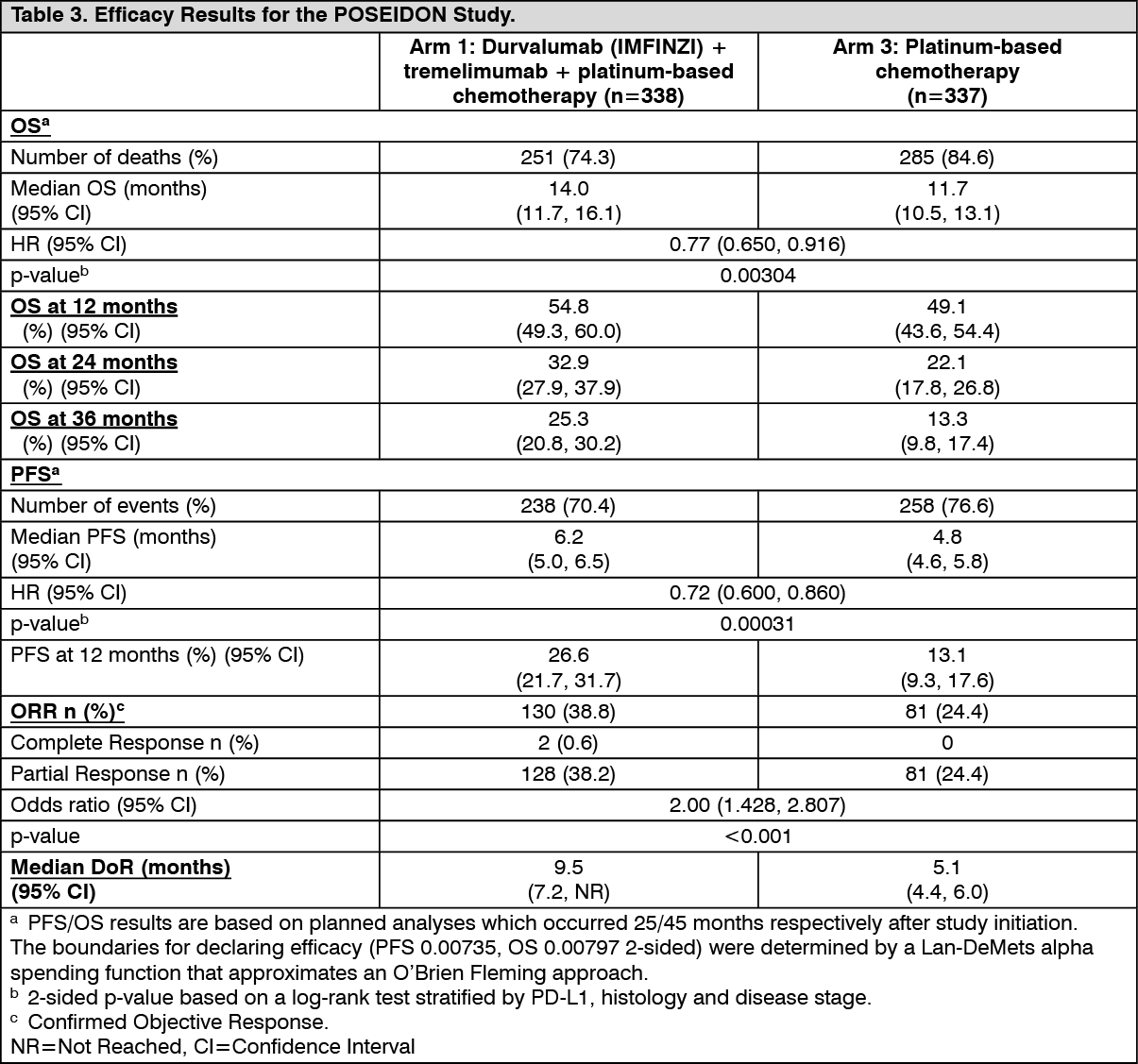 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image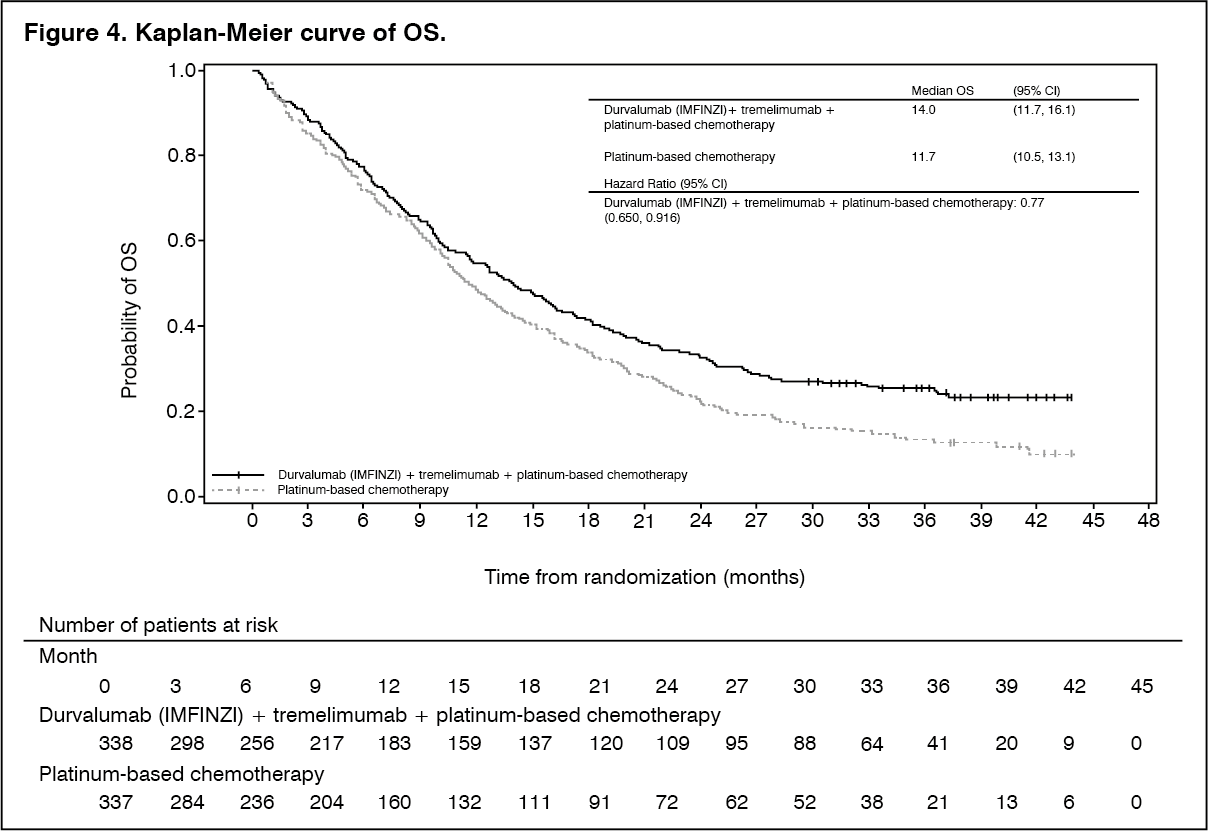 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image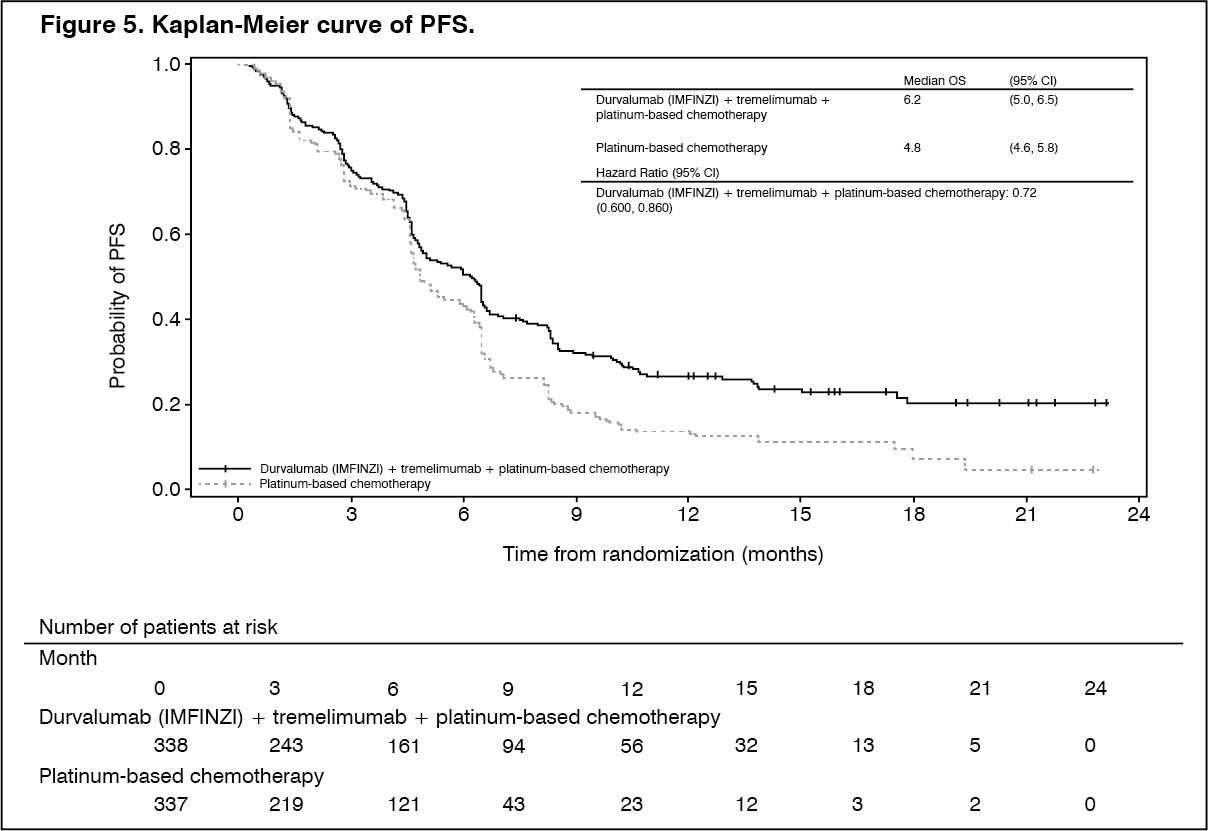 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image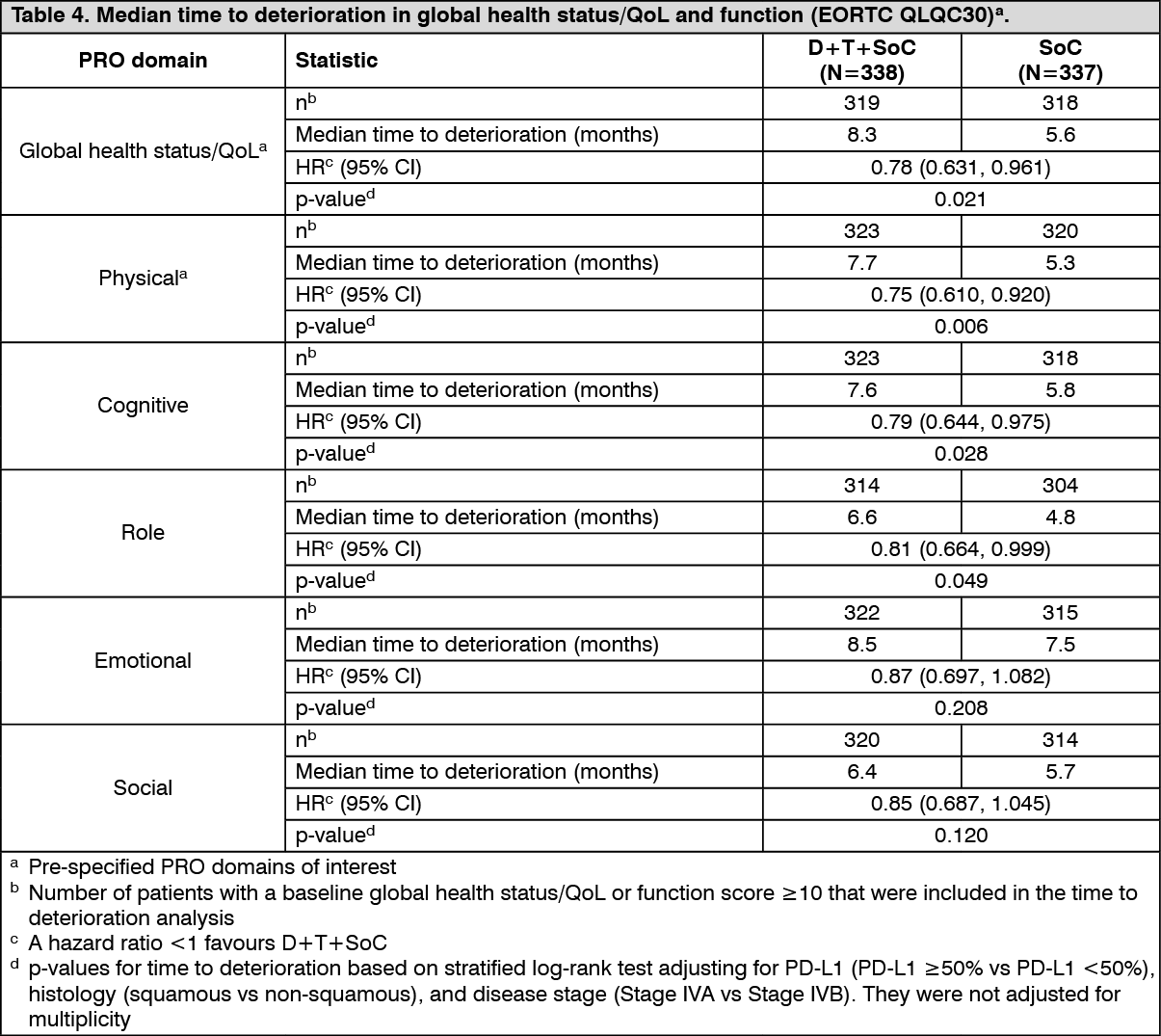 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image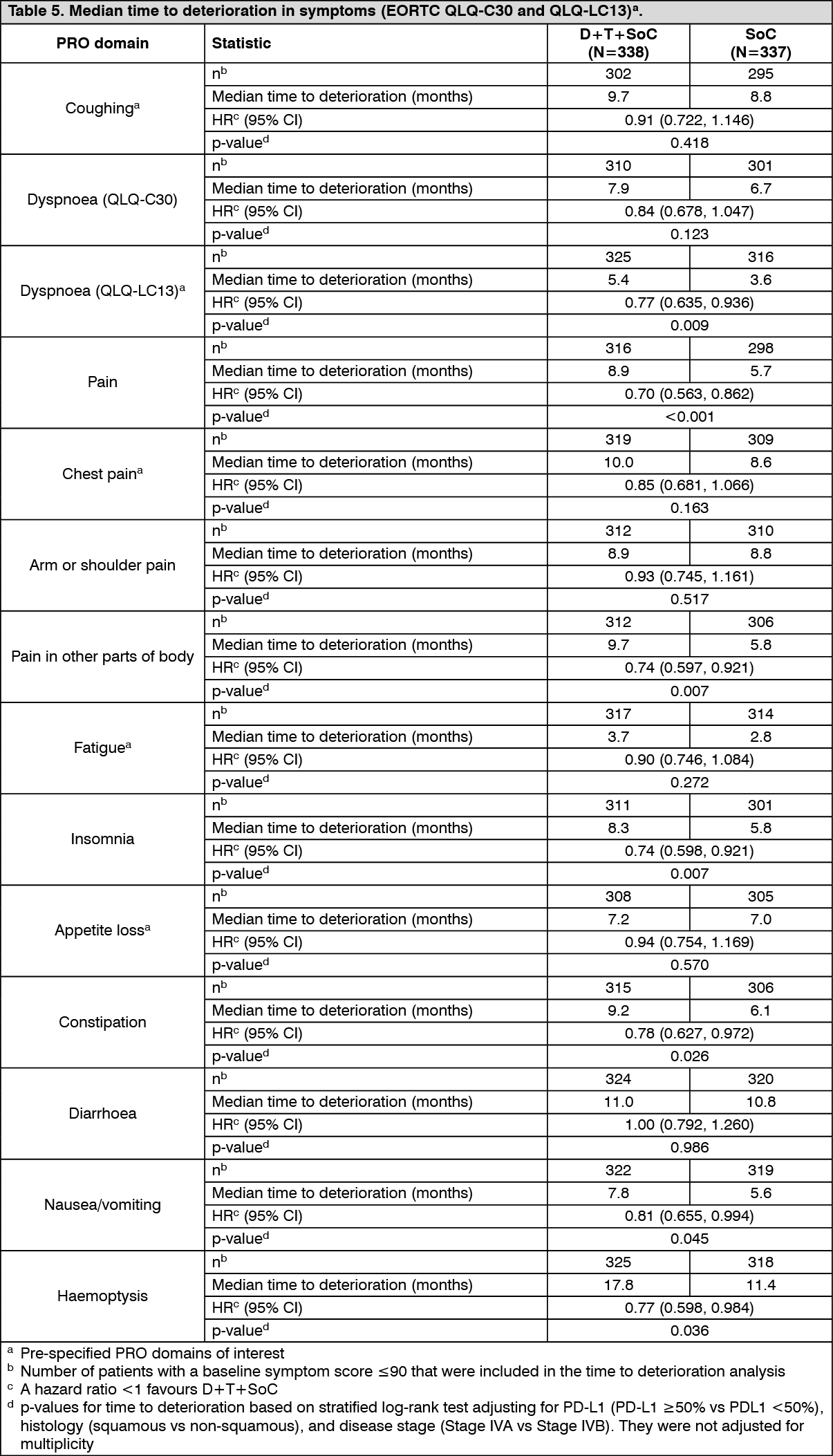 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image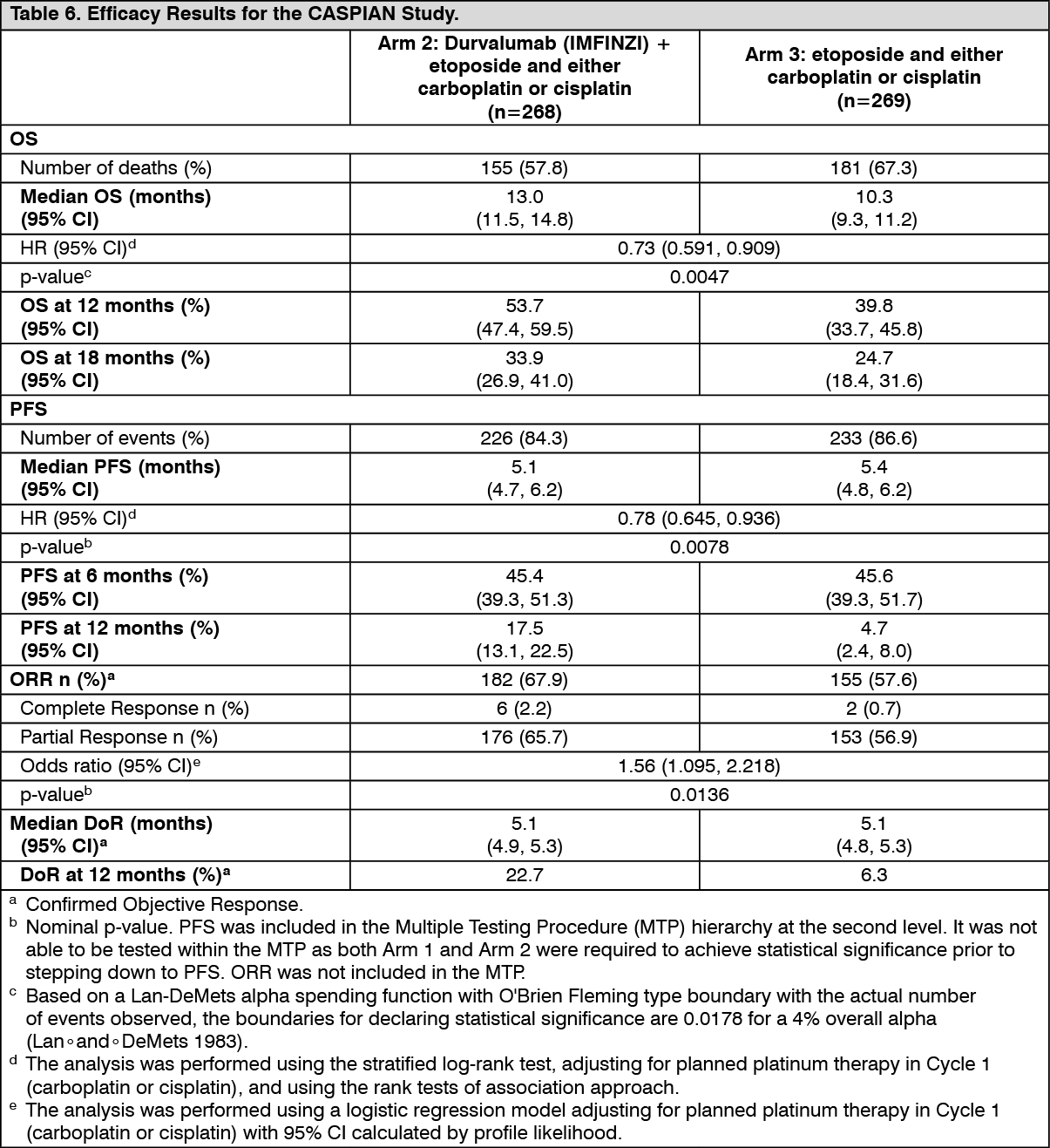 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image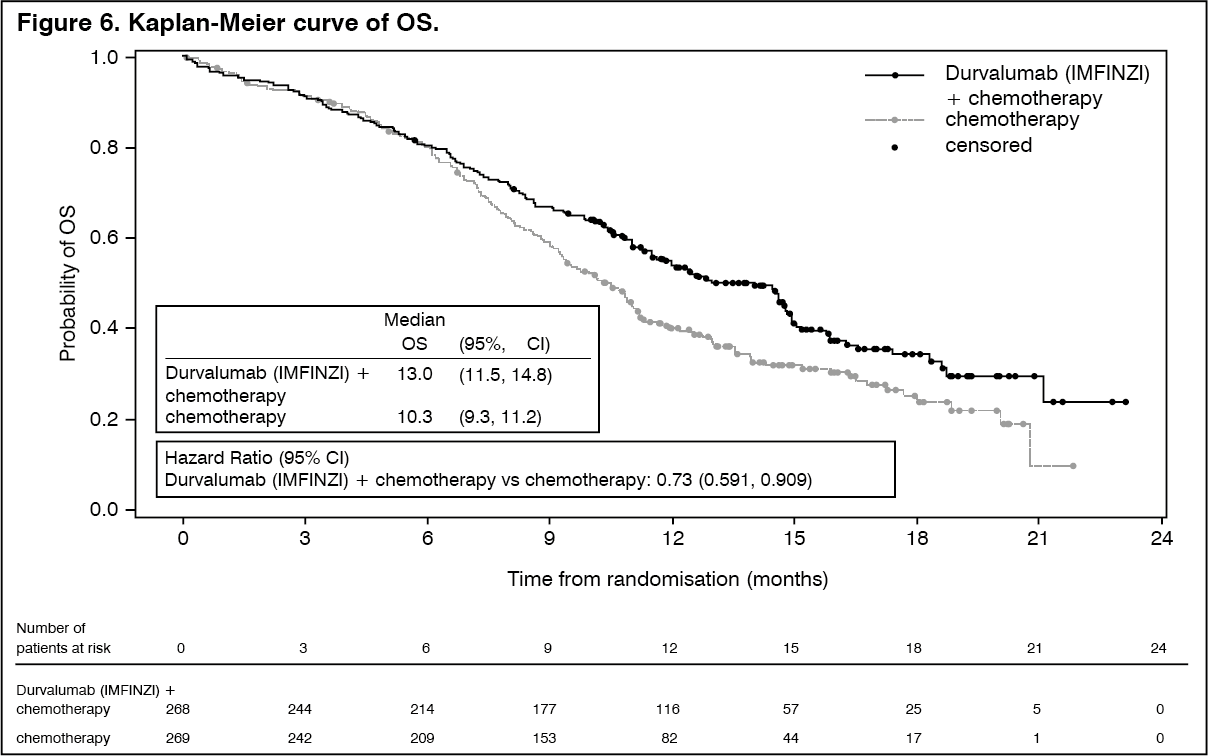 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image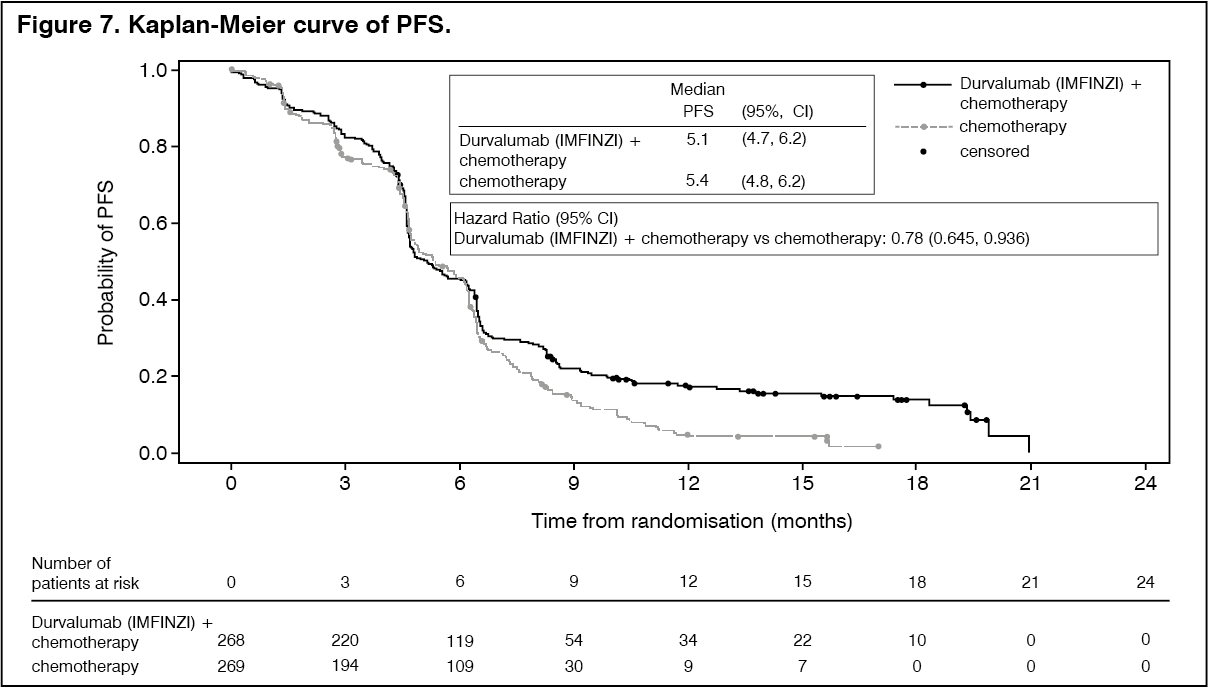 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image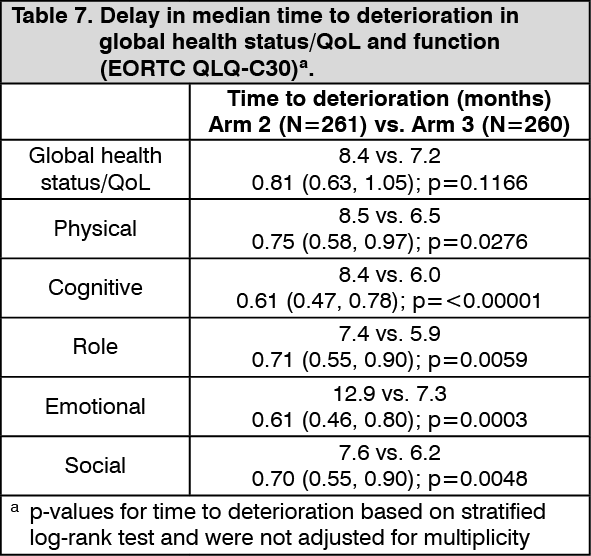 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image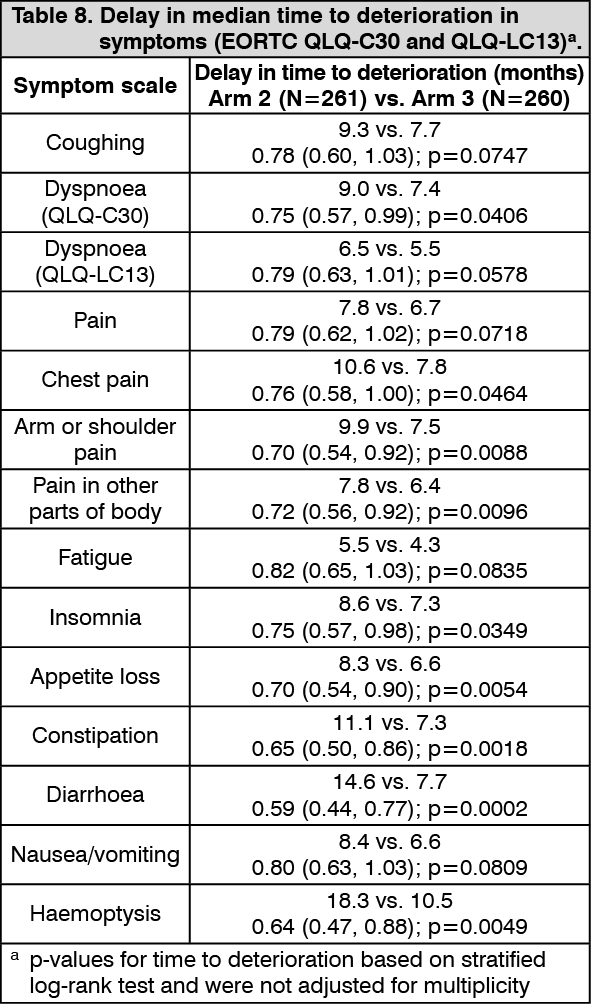 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image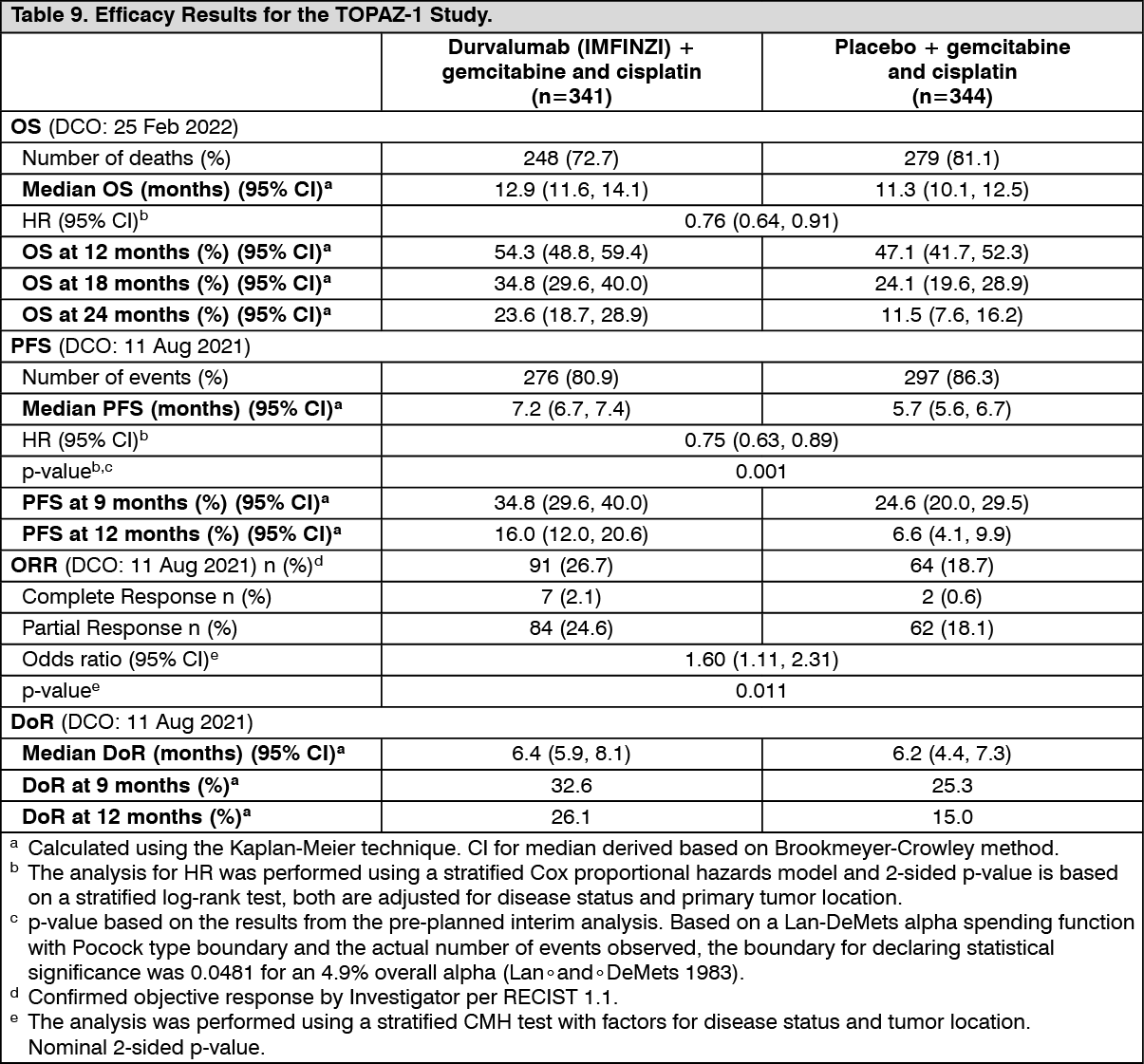 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image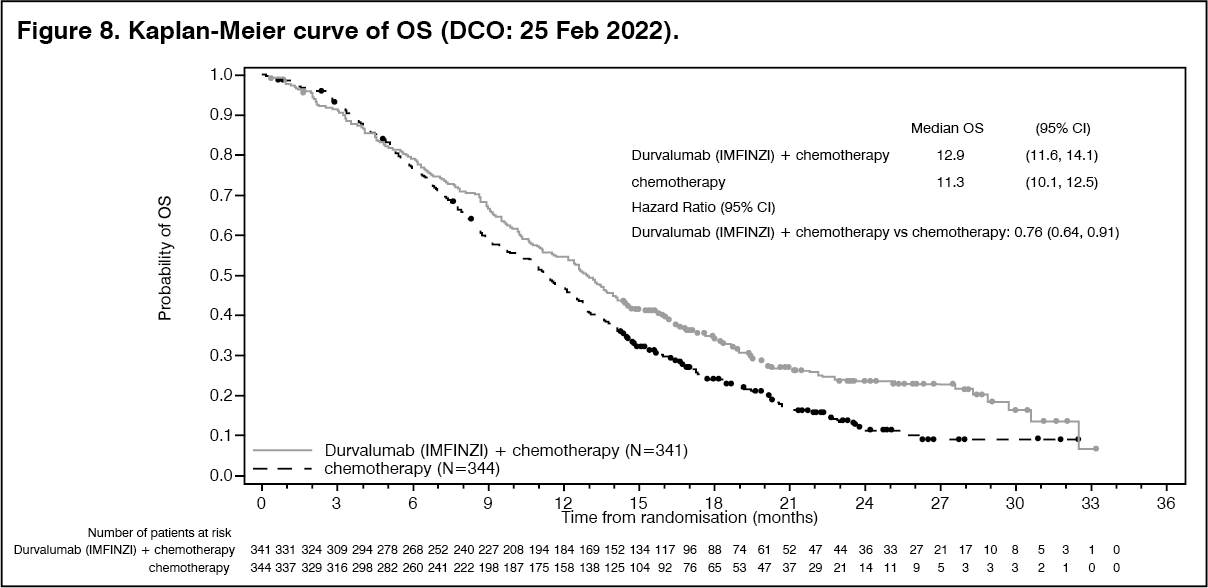 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image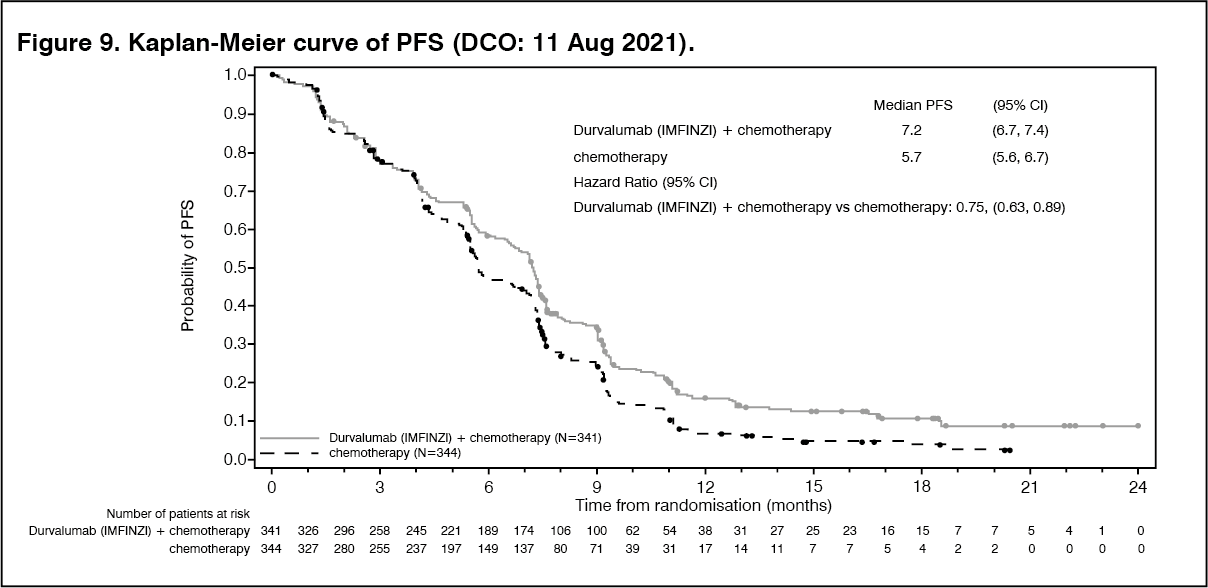 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image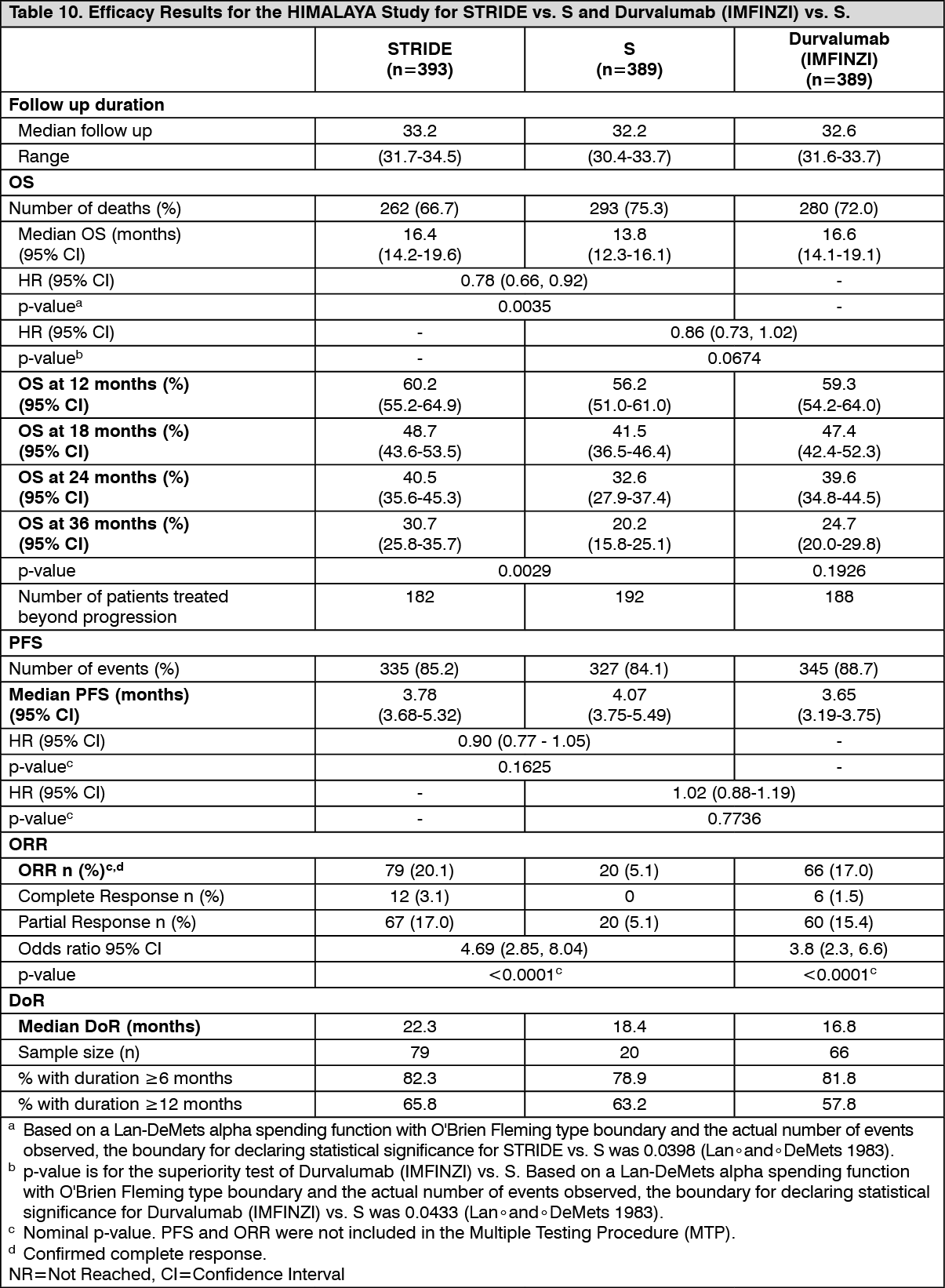 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image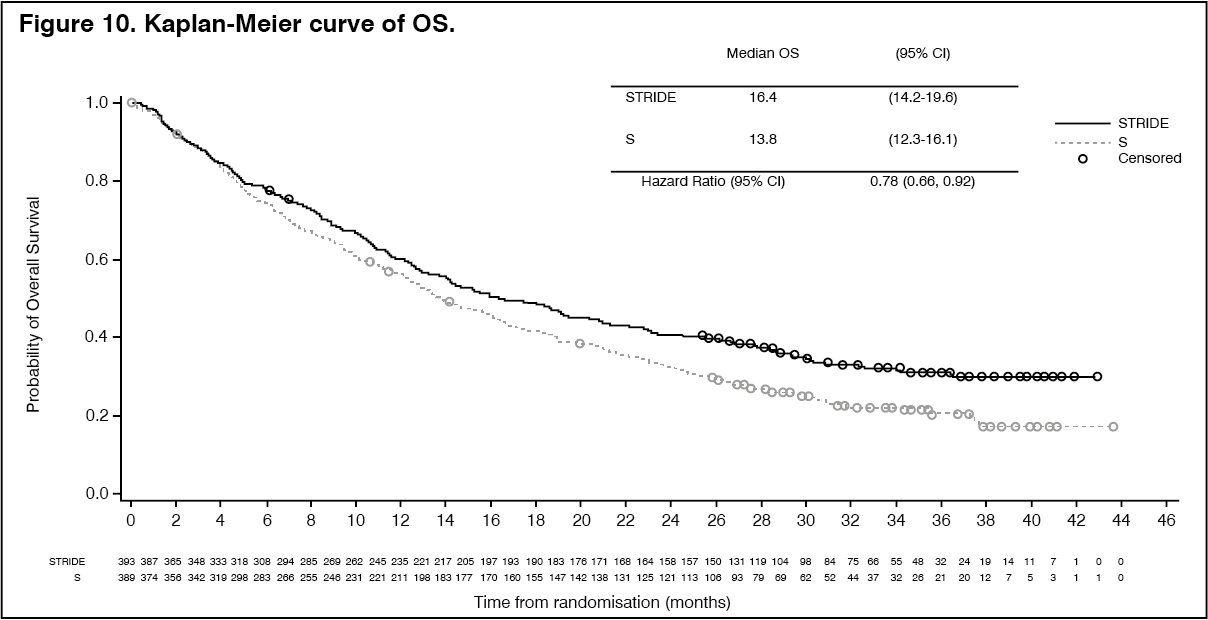 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image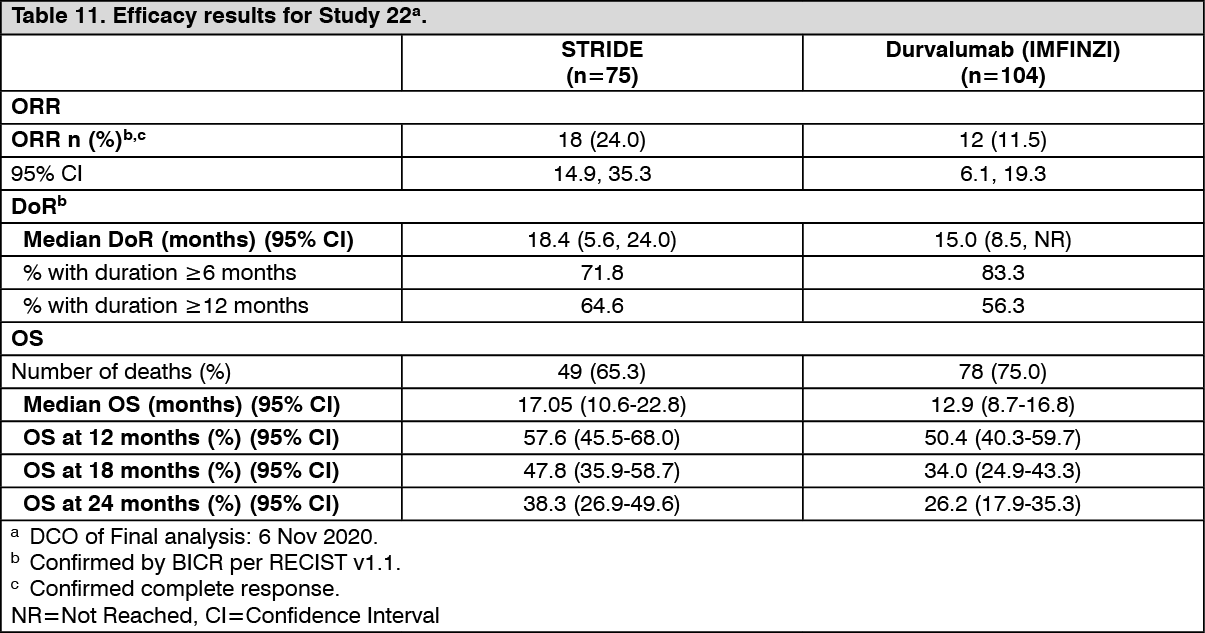 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image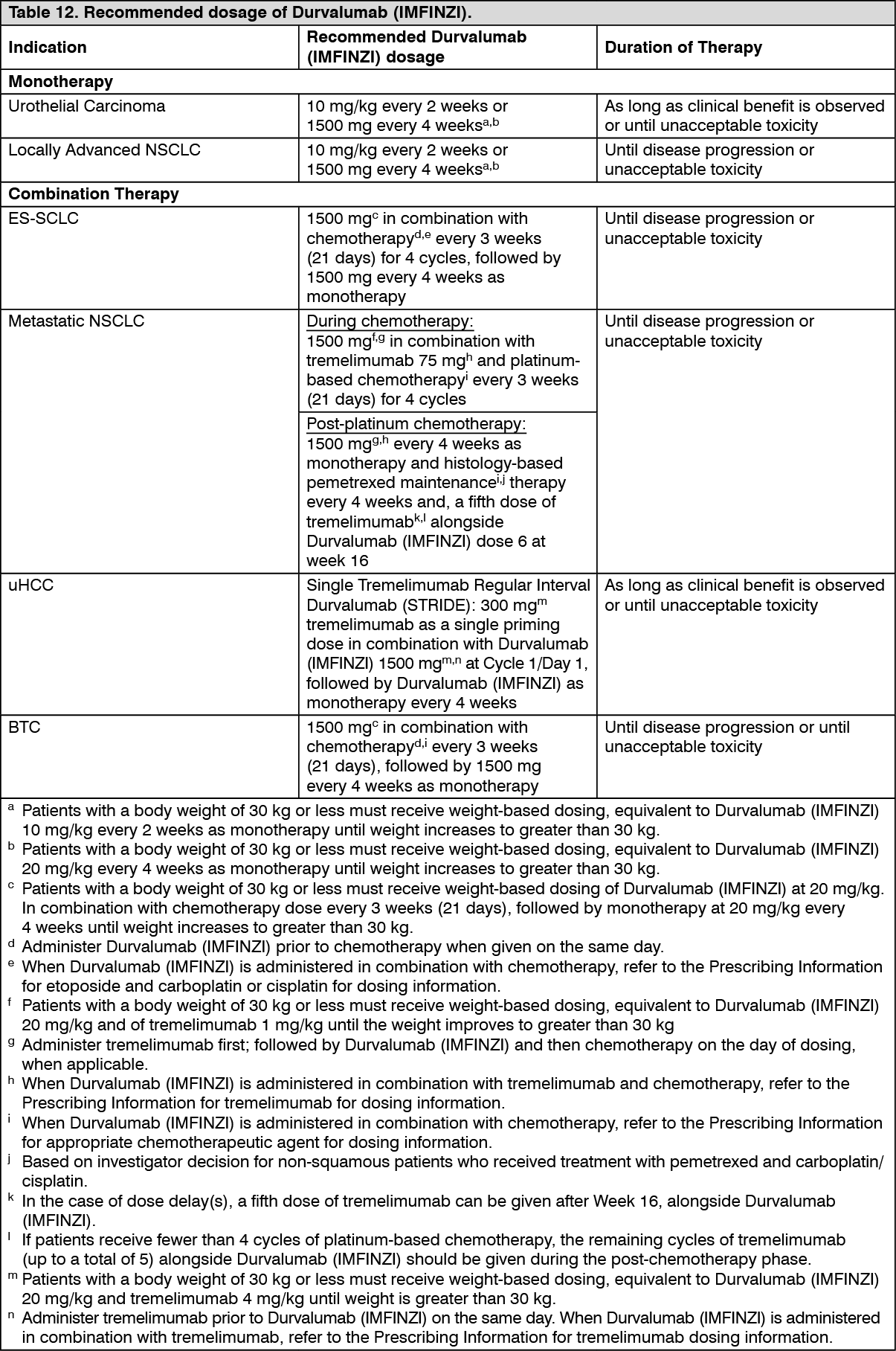 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image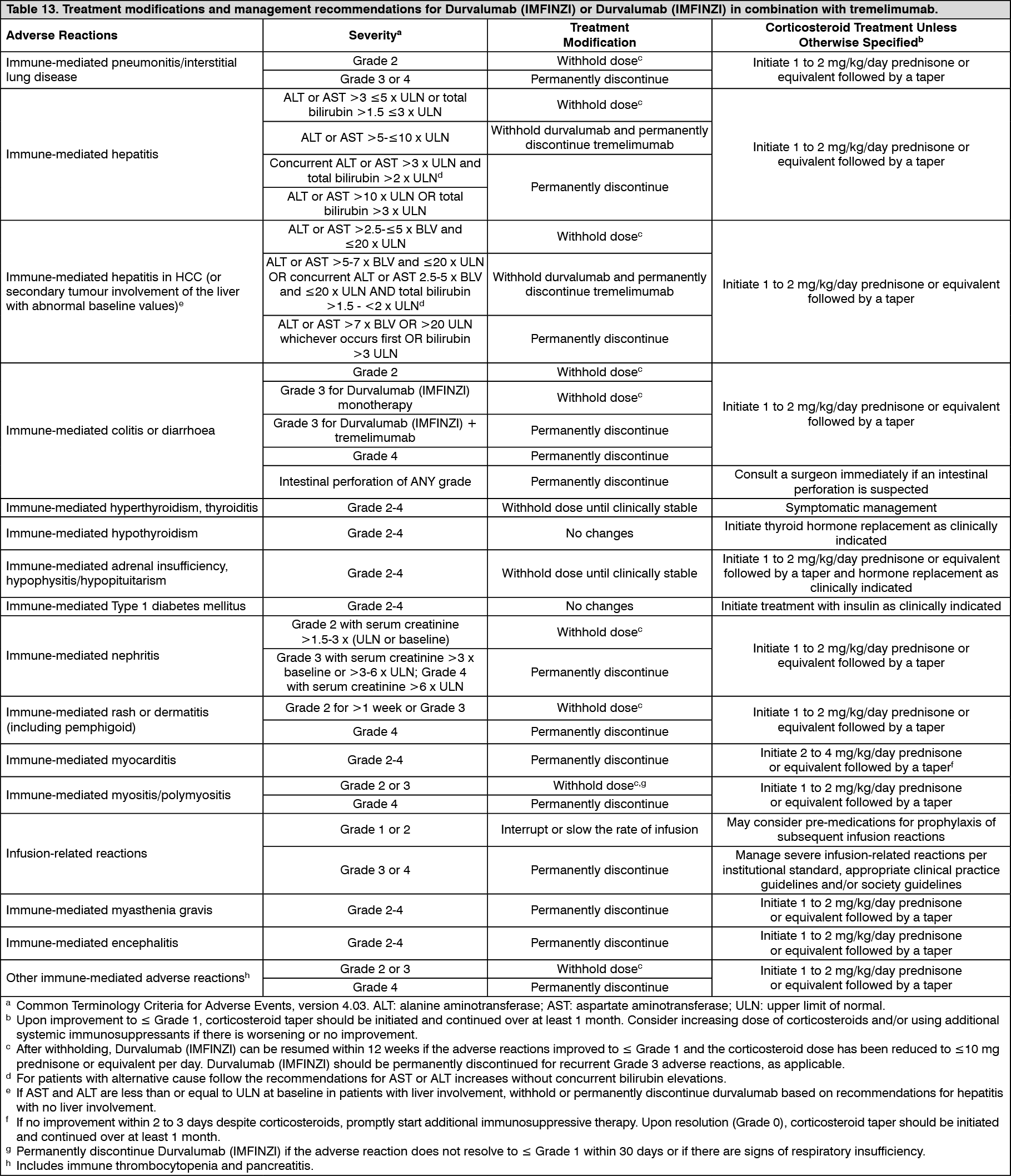 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image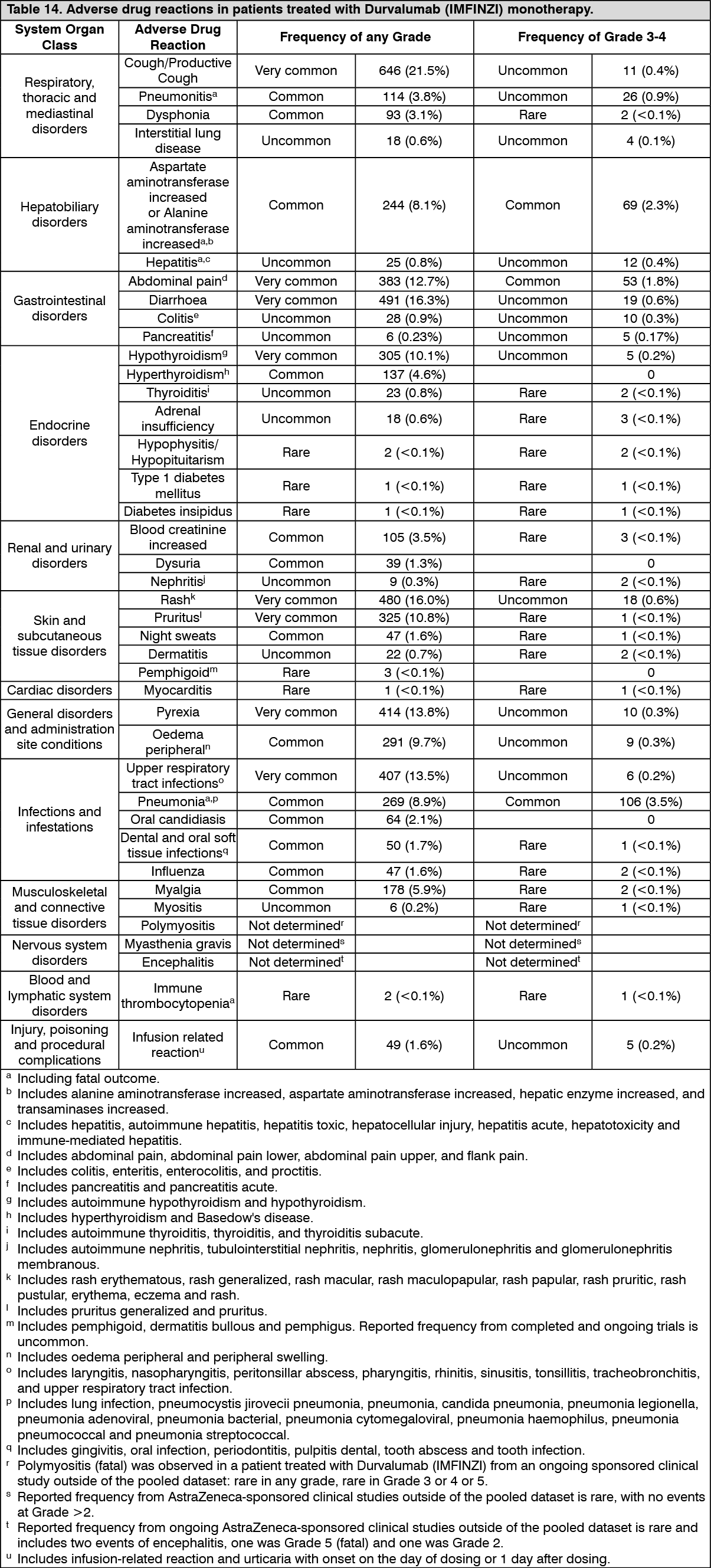 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out