Pharmacology: Pharmacodynamics: Imatinib is protein-tyrosine kinase inhibitor, which potently inhibits the breakpoint cluster region-Abelson (BCR-ABL) tyrosine kinase at the
in vitro, cellular, in vivo levels. The compound selectively inhibits proliferation and induces apoptosis in BCR-ABL positive cell lines as well as fresh leukemic cells from Philadelphia chromosome positive CML and acute lymphoblastic leukemia (ALL) patients. In colony transformation assays using
ex vivo peripheral blood and bone marrow samples, imatinib shows selective inhibition of BCR-ABL positive colonies from CML patients.
In vivo the compound shows anti-tumor activity as a single agent in animal models using BCR-ABL positive tumor cells.
Imatinib is also an inhibitor of the receptor tyrosine kinases for platelet-derived growth factor (PDGF) and stem cell factor (SCF), KIT, and inhibits PDGF- and SCF-mediated cellular events.
In vitro, imatinib inhibits proliferation and induces apoptosis in gastrointestinal stromal tumor (GIST) cells, which express an activating KIT mutation. Constitutive activation of the PDGFR or the ABL protein tyrosine kinases as a consequence of fusion to diverse partner proteins or constitutive production of PDGF have been implicated in the pathogenesis of MDS/MPD, HES/CEL and DFSP. In addition, constitutive activation of KIT or the PDGFR has been implicated in the pathogenesis of SM. Imatinib inhibits signaling and proliferation of cells driven by dysregulated PDGFR, KIT and ABL kinase activity.
Mechanism of action: Imatinib is a small molecule protein-tyrosine kinase inhibitor that potently inhibits the activity of the BCR-ABL tyrosine kinase (TK), as well as several receptor TKs: KIT, the receptor for stem cell factor (SCF) coded for by the KIT proto-oncogene, the discoidin domain receptors (DDR1) and DDR2), the colony stimulating factor receptor (CSF-1R) and the platelet-derived growth factor receptors alpha and beta (PDGFR-alpha and PDGFR-beta). Imatinib can also inhibit cellular events mediated by activation of these receptor kinases.
Clinical Studies: Clinical studies in CML: The effectiveness of imatinib mesylate (Imachron) is based on overall hematological and cytogenetic response rates and progression free survival.
Three large, international, open-label, non-controlled phase II studies were conducted in patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML) in advanced, blast or accelerated phase disease, other Ph+ leukemias or with CML in the chronic phase but failing prior interferon-alpha (IFN) therapy. One large, open-label, multicenter, international randomized phase III study has been conducted in patients with newly diagnosed Ph+ CML. In addition, children have been treated in two phase I studies and one open-label, multicenter, single arm phase II trial.
In all clinical studies 38 to 40% of patients were ≥60 years of age and 10 to 12% of patients were ≥70 years of age.
Chronic phase, newly diagnosed: This phase III study compared treatment with either single-agent imatinib mesylate (Imachron) at 400 mg daily or a combination of 5 MIU/m
2/day IFN and 20 mg/m
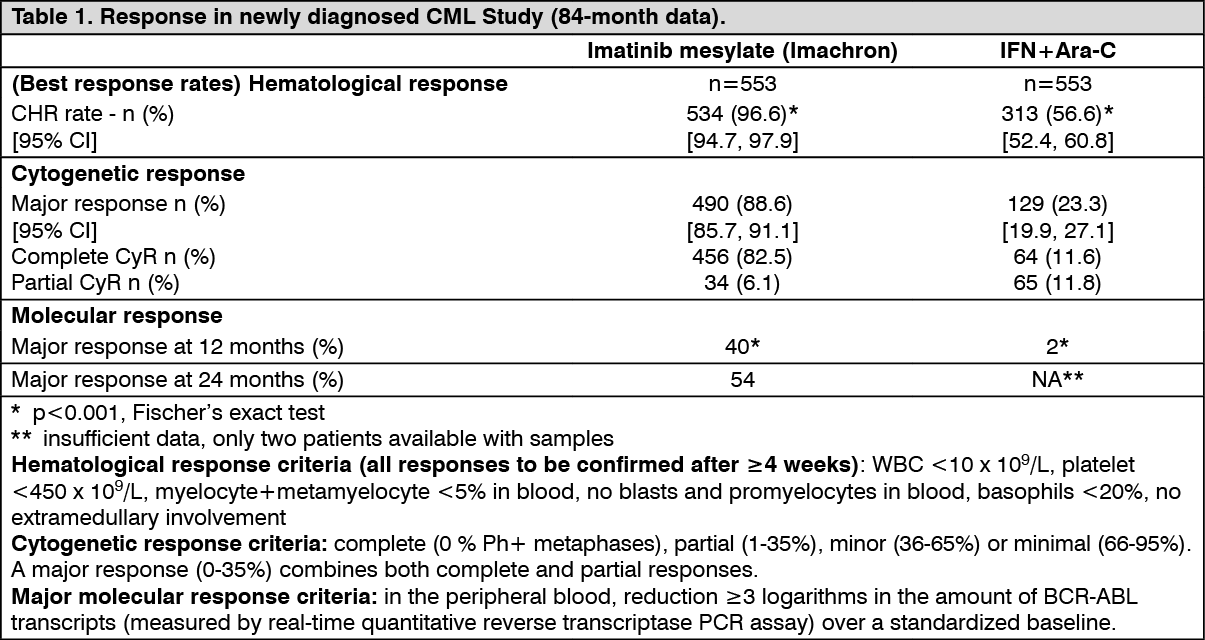
2/day Ara-C, both subcutaneously for 10 days/month. Patients showing lack of response (lack of complete hematological response (CHR) at 6 months, increasing white blood cells (WBC), no major cytogenetic response (MCyR) at 24 months), loss of response (loss of CHR or McyR) or severe intolerance to treatment were allowed to crossover to the alternative treatment arm. A total of 1,106 patients were randomized, 553 to each arm. Median age was 51 years (range 18 to 70 years), with 21.9% of patients ≥60 years of age. 59% males and 41% females; At the 7 year follow-up,, the median duration of first-line treatment was 82 and 8 months in the imatinib mesylate (Imachron) and IFN arm, respectively. The median duration of second-line treatment with imatinib mesylate (Imachron) was 64 months. Overall, in patients receiving first line imatinib mesylate (Imachron), the average daily dose delivered was 406±76 mg. As a consequence of a higher rate of both discontinuations and crossovers, only 2% of patients randomized to IFN are still on first line treatment. In the IFN arm, withdrawal of consent (14%) was the most frequent reason for discontinuation of first line therapy, and the most frequent reason for crossover to the imatinib mesylate (Imachron) arm was severe intolerance to treatment (26%) and progression (14%). The primary efficacy endpoint of the study is progression-free survival. Progression was defined as any of the following event: progression to accelerated phase or blast crisis (AP/BC), death, loss of CHR or MCyR, or in patients not achieving a CHR an increasing WBC despite appropriate therapeutic management. Major cytogenetic response, hematological response, molecular response (evaluation of minimal residual disease), time to accelerated phase or blast crisis and survival are main secondary endpoints. Response data are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
With 7 years follow-up, there were 93 (16.8%) progression events in the imatinib mesylate (Imachron) arm: 37 (6.7%) involving progression to AP/BC, 31 (5.6%) loss of MCyR, 15 (2.7%) loss of CHR or increase in WBC and 10 (1.8%) CML unrelated deaths. In contrast, there were 165 (29.8%) events in the IFN+Ara-C arm of which 130 occurred during first-line treatment with IFN+Ara-C.
The estimated rate of progression-free survival at 84 months is 81.2% with 95% CI (78, 85) in the imatinib mesylate (Imachron) arm and 60.6% (56, 5) in the control arm (p <0.001) (Figure 1). The yearly rates of progression for imatinib mesylate (Imachron) were 3.3% in the 1st year after start of study, 7.5% in the 2nd year and 4.8%, 1.7%, 0.8% 0.3% and 2.0% in the 3rd, 4th, 5th, 6th and 7th year of study respectively.
The estimated rate of patients free of progression to accelerated phase or blast crisis at 84 months was significantly higher in the imatinib mesylate (Imachron) arm compared to the IFN arm (92.5% versus 85.1%, p<0.001). (See Figure 1.)
Click on icon to see table/diagram/image
A total of 71 (12.8%) and 85 (15.4%) patients died in the imatinib mesylate (Imachron) and IFN+Ara-C groups, respectively. At 84 months the estimated overall survival is 86.4% (83, 90) vs. 83.3% (80, 87) in the randomized imatinib mesylate (Imachron) and the IFN+Ara-C groups, respectively (p=0.073, log-rank test). This time-to-event endpoint is strongly affected by the high crossover rate from IFN+Ara-C to imatinib mesylate (Imachron). When censoring the 48 deaths that occurred after BMT, the 84-months survival rates were 89.6 vs 88.1 (p=0.200, log-rank test). Only 31 deaths (before BMT) of the imatinib mesylate (Imachron) patients (5.6%) were attributed to CML, compared to 40 of the IFN+Ara-C patients (7.2%). When only considering these CML-related deaths and censoring any deaths after BMT or due to other reasons, the estimated 84-months survival rates were 93.6% vs. 91.1% (p=0.1, log rank test). In this study, dose escalations were allowed from 400 mg daily to 600 mg daily, then from 600 mg daily to 800 mg daily. After 42 months of follow-up, 11 patients who achieved a CHR at 3 months and a MCyR at 12 months while on a daily dose of 400 mg experienced a confirmed loss (within 4 weeks) of their cytogenetic response. Of these 11 patients, 4 patients escalated up to 800 mg daily, 2 of whom regained a cytogenetic response (1 partial and 1 complete, the latter also achieving a molecular response), while of the 7 patients in whom the dose was not escalated, only one regained a complete cytogenetic response. The percentage of some ADRs was higher in the 40 patients in whom the dose was increased to 800 mg daily compared to the population of patients before dose increase (n=551). These more frequent ADRs included gastrointestinal hemorrhages, conjunctivitis and elevation of transaminases or bilirubin. Other ADRs were reported with lower or equal frequency.
Chronic phase, Interferon-failure: 532 patients were treated at a starting dose of 400 mg. The patients were distributed in three main categories: hematological failure (29%), cytogenetic failure (35%), or intolerance to interferon (36%). Patients had received a median of 14 months of prior IFN therapy at doses ≥25 x 10
6 IU/week and were all in late chronic phase, with a median time from diagnosis of 32 months. The primary efficacy variable of the study was the rate of major cytogenetic response (complete plus partial response, 0 to 35% Ph+ metaphases in the bone marrow).
In this study, 65% of the patients achieved a MCyR, which was complete in 53% of patients CHR was achieved in 95% of patients.
Accelerated phase: 235 patients with accelerated phase disease were enrolled. The first 77 patients were started at 400 mg, and the remaining 158 patients were started at 600 mg.
The primary efficacy variable was the rate of hematological response, reported as either CHR, no evidence of leukemia (i.e. clearance of blasts from the marrow and the blood, but without a full peripheral blood recovery as for complete responses), or return to chronic phase CML. A confirmed hematological response was achieved in 71.5% of patients. Importantly, 27.7% of patients also achieved a MCyR, which was complete in 20.4% of patients. For the patients treated at 600 mg, the current estimates for median progression-free survival and overall survival were 22.9 and 42.5 months, respectively. In a multivariate analysis, a dose of 600 mg was associated with an improved time to progression, independent of platelet count, blood blasts and hemoglobin >10 g/L.
Myeloid blast crisis: 260 patients with myeloid blast crisis were enrolled. 95 (37%) had received prior chemotherapy for treatment of either accelerated phase or blast crisis ("pre-treated patients") whereas 165 (63%) had not ("untreated patients"). The first 37 patients were started at 400 mg, and the remaining 223 patients were started at 600 mg.
The primary efficacy variable was the rate of hematological response, reported as either CHR, no evidence of leukemia, or return to chronic phase CML 31% of patients achieved a hematological response (36% in previously untreated patients and 22% in previously treated patients). The rate of response was also higher in the patients treated at 600 mg (33%) as compared to the patients treated at 400 mg (16%, p=0.0220). The current estimate of the median survival of the previously untreated and treated patients was 7.7 and 4.7 months, respectively.
Pediatric patients: A total of 51 pediatric patients with newly diagnosed and untreated CML in chronic phase were enrolled in an open-label, multicenter, single arm phase II trial, and were treated with imatinib mesylate (Imachron) 340 mg/m
2/day. Imatinib mesylate (Imachron) treatment induced a rapid response in newly diagnosed pediatric CML patients with a CHR of 78% after 8 weeks of therapy and a complete cytogenetic response (CCyR) of 65% (comparable to results in adults) after 3 to 10 months of treatment.
A total of 31 heavily pre-treated pediatric patients (45% with prior BMT and 68% with prior multi-agent chemotherapy) with either chronic phase CML (n=15) or CML in blast crisis or Ph+ ALL (n=16) were enrolled in a dose escalation phase I trial. Patients were treated at doses of imatinib mesylate (Imachron) ranging between 260 mg/m
2/day and 570 mg/m
2/day. Out of 13 patients with CML and cytogenetic data available, 7 (54%) and 4 (31%) achieved a complete and partial cytogenetic response, respectively, for a rate of MCyR of 85%.
Clinical studies in Ph+ ALL: A total of 851 Ph+ ALL patients with either newly diagnosed or relapsed/refractory disease were enrolled in eleven clinical studies, ten of which were uncontrolled and one was randomized. Of the 851 patients, 93 were pediatric patients (including 4 patients older than 18 and younger than 22 years) treated in one open-label, multicenter, non-randomized phase III study.
Newly diagnosed Ph+ ALL: In a controlled study (ADE10) of imatinib mesylate (Imachron) versus chemotherapy induction in 55 newly diagnosed patients aged 55 years and over, imatinib mesylate (Imachron) used as single agent induced a significantly higher rate of complete hematological response than chemotherapy (96.3% vs. 50%; p=0.0001). When salvage therapy with imatinib mesylate (Imachron) was administered in patients who did not respond or who responded poorly to chemotherapy, it resulted in 9 patients (81.8%) out of 11 achieving a complete hematological response. This clinical effect was associated with a higher reduction in BCR-ABL transcripts in the imatinib mesylate (Imachron)-treated patients than in the chemotherapy arm after 2 weeks of therapy (p=0.02). All patients received imatinib mesylate (Imachron) and consolidation chemotherapy after induction and the levels of BCR-ABL transcripts were identical in the two arms at 8 weeks. As expected on the basis of the study design, no difference was observed in remission duration, disease-free survival or overall survival, although patients with complete molecular response and remaining in minimal residual disease had a better outcome in terms of both remission duration (p=0.01) and disease-free survival (p=0.02).
The results observed in a population of 211 newly diagnosed Ph+ ALL patients in four uncontrolled clinical studies (AAU02, ADE04, AJP01 and AUS01) are consistent with the results described previously, Similarly, in two uncontrolled clinical studies (AFR09 and AIT04) 49 newly diagnosed Ph+ ALL patients aged 55 years and over were given imatinib mesylate (Imachron) combined with steroids with or without chemotherapy results are shown in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Pediatric patients: In study I2301, a total of 93 pediatric, adolescent and young adult patients (including 4 patients older than 18 and younger than 22 years) with Ph+ ALL were enrolled in an open-label, multicenter, sequential cohort, non-randomized phase III trial, and were treated with imatinib mesylate (Imachron) (340 mg/m
2/day) in combination with intensive chemotherapy after induction therapy. Imatinib mesylate (Imachron) was administered intermittently in cohorts 1 to 5, with increasing duration and earlier start of imatinib mesylate (Imachron) from cohort to cohort; cohort 1 receiving the lowest intensity and cohort 5 receiving the highest intensity of imatinib mesylate (Imachron) (longest duration in days with continuous daily imatinib mesylate (Imachron) dosing during the first chemotherapy treatment courses). Continuous daily exposure to imatinib mesylate (Imachron) early in the course of treatment in combination with chemotherapy in cohort 5 patients (n=50) improved the 4-year event-free survival (EFS) compared to historical controls (n=120), who received standard chemotherapy without imatinib mesylate (Imachron) (69.6% vs. 31.6%, respectively).
The estimated 4-year OS in Cohort 5 patients was 83.6% compared to 44.8% in the historical controls.
Relapsed/refractory Ph+ ALL: When imatinib mesylate (Imachron) was used as single agent in patients with relapsed/refractory Ph+ ALL, it resulted, in the 66 out of 429 patients evaluable for response, in a hematological response rate of 33% (12% complete) and a major cytogenetic response rate of 23%. The median time to progression in the overall population of 429 patients with relapsed/refractory Ph+ ALL ranged from 1.9 to 3.1 months, and median overall survival in the 409 evaluable patients ranged from 5 to 9 months. In 14 patients, Imatinib mesylate (Imachron) in combination with induction chemotherapy resulted in a complete hematological response rate of 92% in 12 evaluable patients and a major cytogenetic response rate of 100% in 8 evaluable patients. Molecular response was assessed in four patients, and two responded completely.
A total of 14 out of 146 patients were treated with imatinib mesylate (Imachron) 600 mg daily and were evaluable for response; complete hematological response was observed in 5 patients (35%) and major cytogenetic response in 7 patients (50%). Of note, four patients who were treated with a lower dose of imatinib mesylate (Imachron) (400 mg daily) did not respond. In the overall population of 146 patients, median disease-free survival ranged from 2.8 to 3.1 months and median overall survival from 7.4 to 8.9 months.
Clinical studies in MDS/MPD: One open label, multicenter, phase II clinical trial (study B2225) was conducted testing imatinib mesylate (Imachron) in diverse populations of patients suffering from life-threatening diseases associated with ABL, KIT or PDGFR protein tyrosine kinases.
This study included 7 patients with MDS/MPD out of a total of 185 patients treated, 45 of whom had hematological diseases and 140 a variety of solid tumors. These patients were treated with imatinib mesylate (Imachron) 400 mg daily. The ages of the enrolled patients ranged from 20 to 86 years. A further 24 patients with MDS/MPD aged 2 to 79 years were reported in 12 published case reports and a clinical study. These patients also received imatinib mesylate (Imachron) at a dose of 400 mg daily with the exception of three patients who received lower doses. Of the total population of 31 patients treated for MDS/MPD, 14 (45%) achieved a complete hematological response and 9 (29%) a complete cytogenetic response (39% including major and partial responses). Of note, the malignancy carried a translocation, usually involving the chromosome t5q33 or t4q12, resulting in a PDGFR gene re-arrangement in 14 evaluable patients. All of these responded hematologically (12 completely). Cytogenetic response was evaluated in 11 out of 14 patients, all of whom responded (9 patients completely). Only 2 (13%) out of the 16 patients without a translocation associated with PDGFR gene re-arrangement achieved a complete hematological response and one (6%) achieved a major cytogenetic response. A further patient with a PDGFR gene re-arrangement in molecular relapse after bone marrow transplant responded molecularly. Median duration of therapy was 12.9 months (0.8 to 26.7) in the 7 patients treated within study B2225 and ranged between 1 week and more than 18 months in responding patients in the published literature. Results are provided in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Clinical studies in SM: This study B2225 also included 5 patients (aged 49 to 74) with SM, receiving imatinib mesylate (Imachron) 100 mg to 400 mg daily A further 25 patients receiving imatinib mesylate (Imachron) at doses of 100 mg to 400 mg daily, with SM (aged 26 to 85 years) were reported (10 published case reports and case series). Of the total of 30 SM patients, 10 (33%) achieved a complete hematological response and 9 (30%) a partial hematological response (63% overall response rate). Cytogenetic abnormalities were evaluated in 21 of 30 patients treated in the published reports and study B2225. Eight out of 21 patients had a FIP1L1-PDGFR-alpha fusion kinase, which is typically in males with or without eosinophilia. Two patients showed a KIT mutation in the juxta membrane region (one Phe522Cys and one K509I). Sixteen patients had unknown or no detected cytogenetic abnormality. Four patients showed a D816V mutation (the one responder had concomitant CML and SM). The majority of patients in literature with the D816V KIT mutation are not considered sensitive to imatinib mesylate (Imachron). Median duration of therapy was 13 months (range 1.4 to 22.3 months) in 5 patients in study B2225 and ranged between 1 and more than 30 months in responding patients in the literature. Results are provided in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
Clinical studies in HES/CEL: This study, B2225 also included 14 patients (aged 16 to 64) with HES/CEL receiving imatinib mesylate (Imachron) 100 mg to 1,000 mg daily. A further 162 patients receiving imatinib mesylate (Imachron) at doses of 75 mg to 800 mg daily, with HES/CEL (aged 11 to 78 years) were reported (35 published case reports and case series). Of the population of 176 patients treated for HES/CEL, 107 (61%) achieved a complete hematological response and 16 (9%) a partial hematological response (70% overall response rate). Cytogenetic abnormalities were evaluated in 117 of 176 patients treated in the published reports and in study B2225. FIP1L1-PDGFR-alpha fusion kinase was found in 61 of 117 patients. All of these FIP1L1-PDGFR-alpha fusion kinase positive patients achieved a complete hematological response. The FIP1L1-PDGFR-alpha fusion kinase was either negative or unknown in 115 patients, of which 62 (54%) achieved either a complete (n=46) or partial (n=16) hematological response. Results are provided in Table 5. (See Table 5.)
Click on icon to see table/diagram/image
Additionally, improvements in symptomatology and organ dysfunction abnormalities (cardiac, nervous, skin/subcutaneous tissue, respiratory/thoracic/mediastinal, musculoskeletal/connective tissue/vascular, and gastrointestinal organ system) were reported in the case reports.
Clinical studies in unresectable or metastatic GIST: Two open-label, randomized, multinational Phase III studies (SWOG, EORTC) were conducted in patients with unresectable or metastatic malignant gastrointestinal stromal tumors (GIST). A total of 1,640 patients were randomized 1:1 to receive either 400 mg or 800 mg orally q.d. continuously until disease progression or unacceptable toxicity. Crossover was permitted to 800 mg q.d. The studies were designed to compare response rates, progression free survival and overall survival between the dose groups. All patients had a pathologic diagnosis of CD117 positive unresectable and/or metastatic malignant GIST.
The primary objective of the two studies was to evaluate either progression free survival (PFS) with a secondary objective of overall survival (OS) in one study (EORTC) or overall survival with a secondary objective of PFS in the other study (SWOG). A planned analysis of both OS and PFS from the combined datasets from these two studies was conducted. Results from this combined analysis are shown in Table 6. (See Table 6.)
Click on icon to see table/diagram/image
Median follow up for the combined studies was 37.5 months. There was a statistically significant improvement in PFS in the 800 mg treatment group (23.2 vs. 18.9 months in the 400 mg arm (p=0.03). However, there were no observed differences in OS between the treatment groups (p=0.98). The estimated overall PFS for all 1640 patients in these Phase III studies was 21 months and the estimated OS was 48.8 months. Only 5.1% of patients achieved a confirmed complete response and 47.5% achieved a partial response. Treatment at either dose level was generally well tolerated and overall 5.4% of patients withdrew due to toxicity.
One phase II, open-label, randomized multinational study was conducted in patients with unresectable or metastatic GIST. In this study 147 patients were enrolled and randomized to receive either 400 mg or 600 mg orally once daily for up to 36 months.
The primary evidence of efficacy was based on objective response rates. Response characterization was based on Southwestern Oncology Group (SWOG) criteria. In this study, 83% of the patients achieved either a complete response, partial response or stable disease. Results are provided in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
There were no differences in response rates between the two dose groups (median follow-up 31 months). Median time to response was 13 weeks. Median time to treatment failure in responders was 122 weeks, while in the overall study population it was 84 weeks. The median overall survival has not been reached. The Kaplan-Meier estimate for survival after 36-month follow-up is 68% (Figure 2). Additionally, there is no difference in survival between patients achieving stable disease and partial response (Figure 3). (See Figure 2 and Figure 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Clinical studies in adjuvant GIST: In the adjuvant setting, imatinib mesylate (Imachron) was investigated in a multicenter, double-blind, long-term, placebo controlled phase III study (Z9001) involving 713 patients. The primary endpoint of the study was recurrence free survival (RFS) defined as the time from date of randomization to the date of recurrence or death from any cause.
Imatinib mesylate (Imachron) prolonged significantly RFS with 75% of patients being recurrence-free at 38 months in the imatinib mesylate (Imachron) group vs 20 months in the placebo group (95% CIs, [30 non-estimable]; [14 non-estimable], respectively); (hazard ratio = 0.398 [0.259 to 0.610], p<0.0001). At one year the overall RFS was significantly better for imatinib mesylate (Imachron) (97.7%) vs. placebo (82.3%), (p<0.0001) therefore reducing the risk of recurrence by approximately 89% as compared with placebo (hazard ratio = 0.113 [0.049 to 0.264]).
A second open label phase III study (SSG XVIII/AIO) compared 400 mg/day imatinib mesylate (Imachron) 12 months treatment vs. 36 months treatment in patients after surgical resection of GIST and one of the following: tumor diameter >5 cm and mitotic count >5/50 high power fields (HPF); or tumor diameter >10 cm and any mitotic count or tumor of any size with mitotic count >10/50 HPF or tumors ruptured into the peritoneal cavity. There were a total of 397 patients consented and randomized to the study (199 patients on 12 month arm and 198 patients on 36 month arm), median age was 61 years (range 22 to 84 years). The median time of follow-up was 54 months (from date of randomization to data cut-off), with a total of 83 months between the first patient randomized and the cut-off date.
The primary endpoint of the study was recurrence free survival (RFS) defined as the time from date of randomization to the date of recurrence or death from any cause.
Thirty-six (36) months of imatinib mesylate (Imachron) treatment significantly prolonged RFS compared to12 months of imatinib mesylate (Imachron) treatment (with overall Hazard Ratio (HR)=0.46 [0.32, 0.65], p<0.0001 and a HR of 0.42 [0.28, 0.61] beyond month 12) (Table 8, Figure 4). There were 84 (42%) and 50 (25%) total RFS events for the 12-months and 36 months arms respectively.
In addition, thirty-six (36) months of imatinib mesylate (Imachron) treatment significantly prolonged overall survival (OS) compared to 12 months of imatinib mesylate (Imachron) treatment (HR=0.45 [0.22, 0.89], p=0.0187) (Table 8, Figure 5). The total number of deaths were 25 for the 12-months treatment arm and 12 for the 36-months treatment arm. (See Table 8, Figure 4 and Figure 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Clinical studies in DFSP: One open label, multicenter, phase II clinical trial (study B2225) was conducted testing imatinib mesylate (Imachron) in a diverse populations of patients suffering from life-threatening diseases associated with ABL, KIT or PDGFR protein tyrosine kinases. This study included 12 patients with DFSP out of a total of 185 patients, 45 of whom had hematological diseases and 140 a variety of solid tumors. The primary evidence of efficacy for patients in the solid tumor group was based on objective response rates. The solid tumor population was treated with imatinib mesylate (Imachron) 800 mg daily. The age of the DFSP patients ranged from 23 to 75 years; DFSP was metastatic, locally recurrent following initial resective surgery and not considered amenable to further resective surgery at the time of study entry. A further 6 DFSP patients treated with imatinib mesylate (Imachron) are reported in 5 published case reports, their ages ranging from 18 months to 49 years. The total population treated for DFSP comprises 18 patients, 8 of them with metastatic disease. The adult patients reported in the published literature were treated with either 400 mg (4 cases) or 800 mg (1 case) imatinib mesylate (Imachron) daily. The pediatric patient received 400 mg/m
2/daily, subsequently increased to 520 mg/m
2/daily. Responses to treatment are described in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
Twelve of these 18 patients either achieved a complete response (7 patients) or were made disease free by surgery after a partial response (5 patients, including one child) for a total complete response rate of 67%. A further 3 patients achieved a partial response, for an overall response rate of 83%. Of the 8 patients with metastatic disease, five responded (62%), three of them completely (37%). The median duration of therapy in study B2225 was 6.2 months, with a maximum duration of 24.3 months, while in the published literature it ranged between 4 weeks and more than 20 months.
Clinical studies in hepatic insufficiency: In a study of patients with varying degrees of hepatic dysfunction (mild, moderate and severe see Table 10 as follows for liver function classification), the mean exposure to imatinib (dose normalized AUC) did not increase compared to patients with normal liver function. In this study, 500 mg daily was safely used in patients with mild liver dysfunction and 300 mg daily was used in other patients. Although only a 300 mg daily dose was used in patients with moderate and severe liver dysfunction, pharmacokinetic analysis projects that 400 mg can be used safely (see Dosage & Administration, Precautions, Adverse Reactions and Pharmacology: Pharmacokinetics under Actions). (See Table 10.)
Click on icon to see table/diagram/image
Clinical studies in renal insufficiency: In a study of patients with varying degrees of renal dysfunction (mild, moderate and severe see Table 11 as follows for renal function classification), the mean exposure to imatinib (dose normalized AUC) increased 1.5- to 2-fold compared to patients with normal renal function, which corresponded to an elevated plasma level of AGP, a protein to which imatinib binds strongly. No correlation between imatinib exposure and the severity of renal deficiency was observed. In this study, 800 mg daily was safely used in patients with mild renal dysfunction and 600 mg daily was used in moderate renal dysfunction. The 800 mg dose was not tested in patients with moderate renal dysfunction due to the limited number of patients enrolled. Similarly, only 2 patients with severe renal dysfunction were enrolled at the low (100 mg) dose, and no higher doses were tested. No patients on hemodialysis were enrolled in the study. Literature data showed that a daily dose of 400 mg was well tolerated in a patient with end-stage renal disease on hemodialysis. The PK plasma exposure in this patient fell within the range of values of imatinib and its metabolite CGP74588 observed in patients with normal renal function. Dialysis was not found to intervene with the plasma kinetics of imatinib. Since renal excretion represents a minor elimination pathway for imatinib, patients with severe renal insufficiency and on dialysis could receive treatment at the 400 mg starting dose. However, in these patients caution is recommended. The dose can be reduced if not tolerated, or increased for lack of efficacy (see Dosage & Administration, Precautions, Adverse Reactions and Pharmacology: Pharmacokinetics under Actions). (See Table 11.)
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics of imatinib mesylate (Imachron) have been evaluated over a dosage range of 25 to 1,000 mg. Plasma pharmacokinetic profiles were analyzed on day 1 and on either day 7 or day 28, by which time plasma concentrations had reached steady state.
Absorption: Mean absolute bioavailability for the capsule formulation imatinib is 98%. The coefficient of variation for plasma imatinib AUC is in the range of 40% to 60% after an oral dose. When given with a high fat meal, the rate of absorption of imatinib was minimally reduced (11% decrease in C
max and prolongation of t
max by 1.5 h), with a small reduction in AUC (7.4%) compared to fasting conditions.
Distribution: At clinically relevant concentrations of imatinib, binding to plasma proteins was approximately 95% on the basis of
in vitro experiments, mostly to albumin and alpha-acid-glycoprotein, with little binding to lipoprotein.
Metabolism: The main circulating metabolite in humans is the N-demethylated piperazine derivative (CGP71588), which shows similar
in vitro potency as the parent compound.
The plasma AUC for this metabolite was found to be only 16% of the AUC for imatinib. The plasma protein binding of the N-demethylated metabolite is similar to that of the parent compound.
Elimination: Based on the recovery of compound(s) after an oral
14C-labelled dose of imatinib, approximately 81% of the dose was eliminated within 7 days in feces (68% of dose) and urine (13% of dose). Unchanged imatinib accounted for 25% of the dose (5% urine, 20% feces), the remainder being metabolites.
Plasma pharmacokinetics: Following oral administration in healthy volunteers, the t
1/2 was approximately 18 h, suggesting that once-daily dosing is appropriate. The increase in mean AUC with increasing dose was linear and dose proportional in the range of 25 to 1,000 mg imatinib after oral administration. There was no change in the kinetics of imatinib on repeated dosing, and accumulation was 1.5 to 2.5 fold at steady state when dosed once daily.
Population pharmacokinetics: Based on population pharmacokinetic analysis, there was a small effect of age on the volume of distribution (12% increase in patients >65 years old). This change is not thought to be clinically significant. The effect of body weight on the clearance of imatinib is such that for a patient weighing 50 kg the mean clearance is expected to be 8.5 L/h, while for a patient weighing 100 kg the clearance will rise to 11.8 L/h. These changes are not considered sufficient to warrant dose adjustment based on kg bodyweight. There is no effect of gender on the kinetics of imatinib.
Further population PK analysis in the phase III study in newly diagnosed CML patients showed that the effect of covariates and co-medications on both clearance and volume of distribution appears to be small and is not sufficiently pronounced to warrant dose adjustment.
Pharmacokinetics in children: As in adult patients, imatinib was rapidly absorbed after oral administration in pediatric patients in both phase I and phase II studies. Dosing in children at 260 and 340 mg/m
2 achieved the same exposure, respectively, as doses of 400 mg and 600 mg in adult patients. The comparison of AUC
(0-24) on Day 8 and Day 1 at 340 mg/m
2 dose level revealed a 1.7 fold drug accumulation after repeated once daily dosing.
Based on pooled population pharmacokinetic analysis in pediatric patients with hematological disorders (CML, Ph+ALL, or other hematological disorders treated with imatinib), clearance of imatinib increases with increasing body surface area (BSA). After correcting for the BSA effect, other demographics such as age, body weight and body mass index did not have clinically significant effects on the exposure of imatinib. The analysis confirmed that exposure of imatinib in pediatric patients receiving 260 mg/m
2 once daily (not exceeding 400 mg once daily) or 340 mg/m
2 once daily (not exceeding 600 mg once daily) were similar to those in adult patients who received imatinib 400 mg or 600 mg once daily.
Organ function impairment: Imatinib and its metabolites are not excreted via the kidney to a significant extent. Patients with mild and moderate impairment of renal function appear to have a higher plasma exposure than patients with normal renal function. The increase is approximately 1.5 to 2 fold, corresponding to a 1.5 fold elevation of plasma AGP, to which imatinib binds strongly. The free drug clearance of imatinib is probably similar between patients with renal impairment and those with normal renal function, since renal excretion represents only a minor elimination pathway for imatinib (see Dosage & Administration, Precautions and Pharmacology: Pharmacodynamics under Actions).
Although the results of pharmacokinetic analysis showed that there is considerable inter-subject variation, the mean exposure to imatinib did not increase in patients with varying degrees of liver dysfunction as compared to patients with normal liver function (see Dosage & Administration, Precautions, Adverse Reactions, Pharmacology: Pharmacodynamics and Pharmacokinetics under Actions).
Toxicology: Preclinical Safety Data: Imatinib has been evaluated in safety pharmacology, repeated dose toxicity, genotoxicity and reproductive toxicity studies. Target organs associated with the pharmacological action of imatinib include bone marrow, peripheral blood, lymphoid tissues, gonads and gastrointestinal tract. Other target organs include the liver and the kidney.
Imatinib was embryotoxic and teratogenic in rats. Fertility was not affected in the preclinical fertility and early embryonic development study although lower testes and epididymal weights as well as a reduced number of motile sperm were observed in the high dose males rats. In the preclinical pre- and postnatal study in rats, fertility in the first generation offspring was also not affected by imatinib mesylate (Imachron).
No new target organs were identified in the rat juvenile development toxicology study (day 10 to 70 post-partum). In the juvenile toxicology study, transitory effects upon growth and delay in vaginal opening and preputial separation were observed at approximately 0.3 to 2 times the average pediatric exposure at the highest recommended dose of 340 mg/m
2. Also, mortality was observed in juvenile animals (around weaning phase) at approximately 2-times the average pediatric exposure at the highest recommended dose of 340 mg/m
2.
In the 2 year rat carcinogenicity study administration of imatinib at 15, 30 and 60 mg/kg/day resulted in a statistically significant reduction in the longevity of males at 60 mg/kg/day and females at ≥30 mg/kg/day. Histopathological examination of decedents revealed cardiomyopathy (both sexes), chronic progressive nephropathy (females) and preputial gland papilloma as principal causes of death or reasons for sacrifice. Target organs for neoplastic changes were the kidneys, urinary bladder, urethra, preputial and clitoral gland, small intestine, parathyroid glands, adrenal glands and non-glandular stomach. The no observed effect levels (NOEL) for the various target organs with neoplastic lesions were established as follows: 30 mg/kg/day for the kidneys, urinary bladder, urethra, small intestine, parathyroid glands, adrenal glands and non-glandular stomach, and 15 mg/kg/day for the preputial and clitoral gland.
The papilloma/carcinoma of the preputial/clitoral gland were noted at 30 and 60 mg/kg/day, representing approximately 0.5 to 4 or 0.3 to 2.4 times the human daily exposure (based on AUC) at 400 mg/day or 800 mg/day, respectively, and 0.4 to 3.0 times the daily exposure in children (based on AUC) at 340 mg/m
2. The renal adenoma/carcinoma, the urinary bladder and urethra papilloma, the small intestine adenocarcinomas, the parathyroid glands adenomas, the benign and malignant medullary tumors of the adrenal glands and the non-glandular stomach papillomas/carcinomas were noted at 60 mg/kg/day.
The relevance of these findings in the rat carcinogenicity study for humans is not known. An analysis of the safety data from clinical trials and spontaneous adverse event reports did not provide evidence of an increase in overall incidence of malignancies in patients treated with imatinib mesylate (Imachron) compared to that of the general population.
Non-neoplastic lesions not identified in earlier preclinical studies were the cardiovascular system, pancreas, endocrine organs and teeth. The most important changes included cardiac hypertrophy and dilatation, leading to signs of cardiac insufficiency in some animals.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image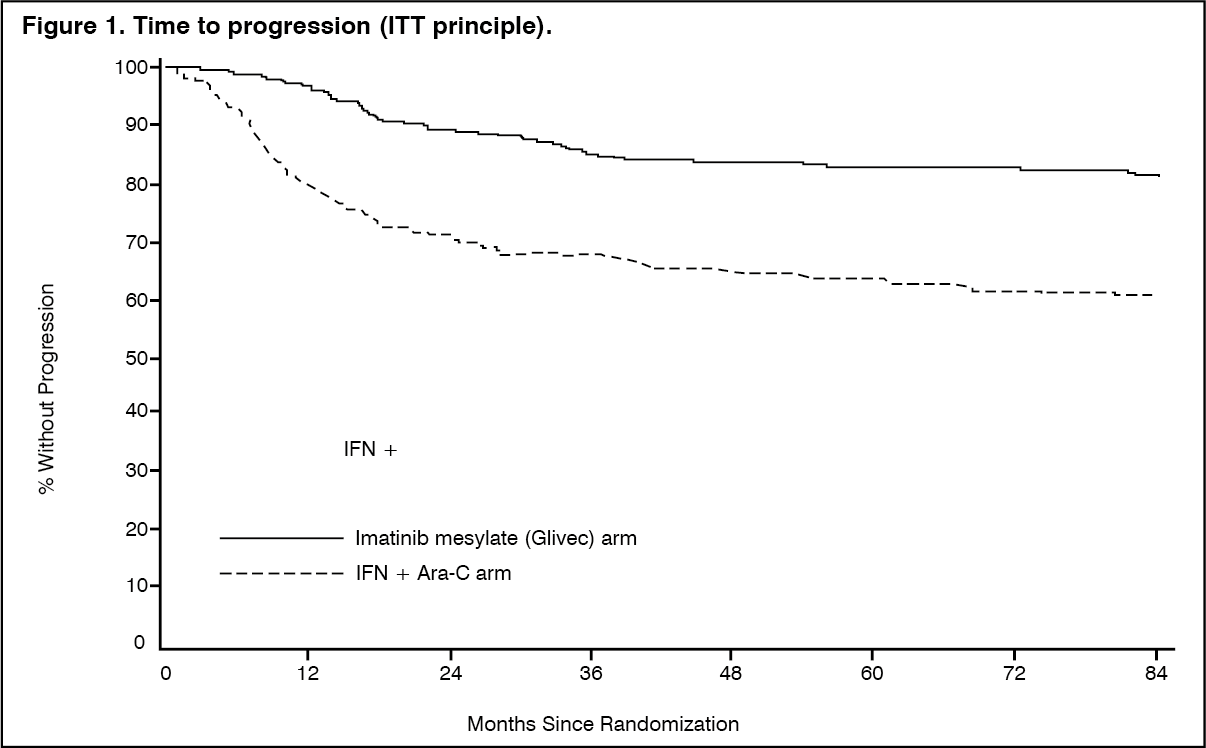 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image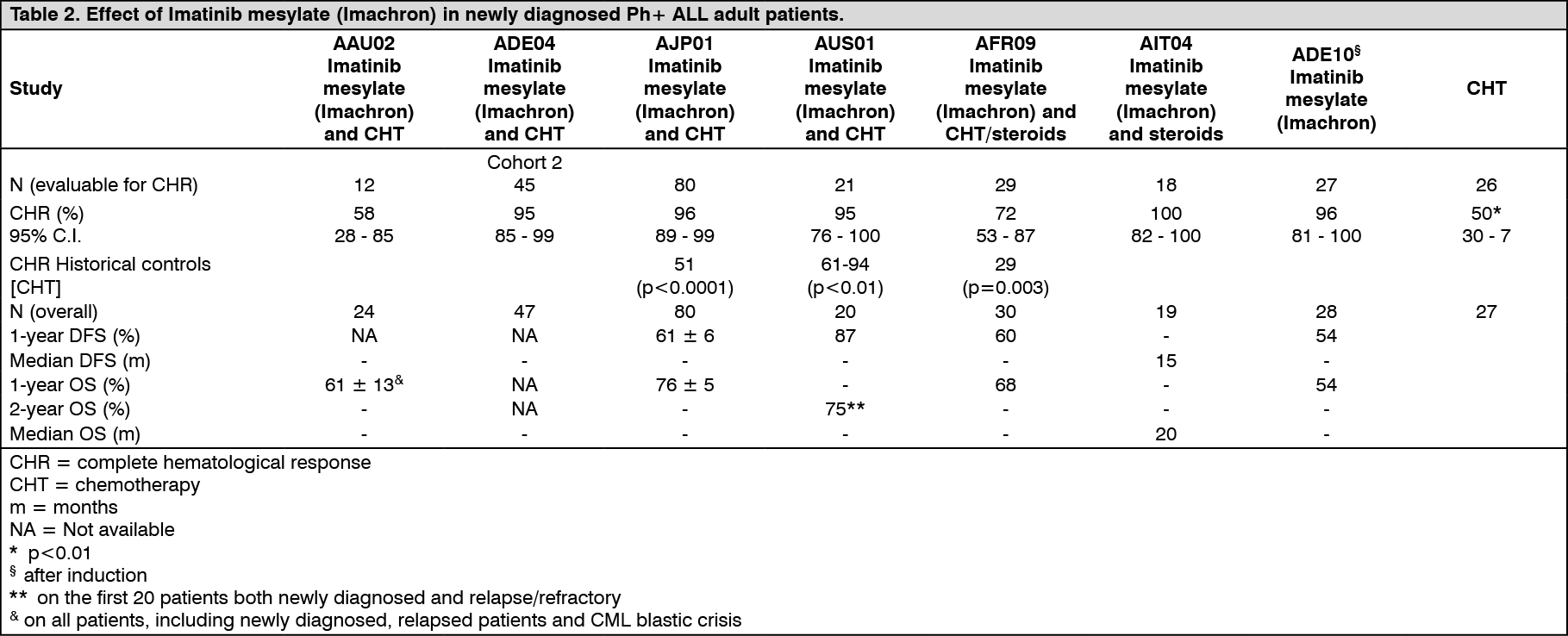 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image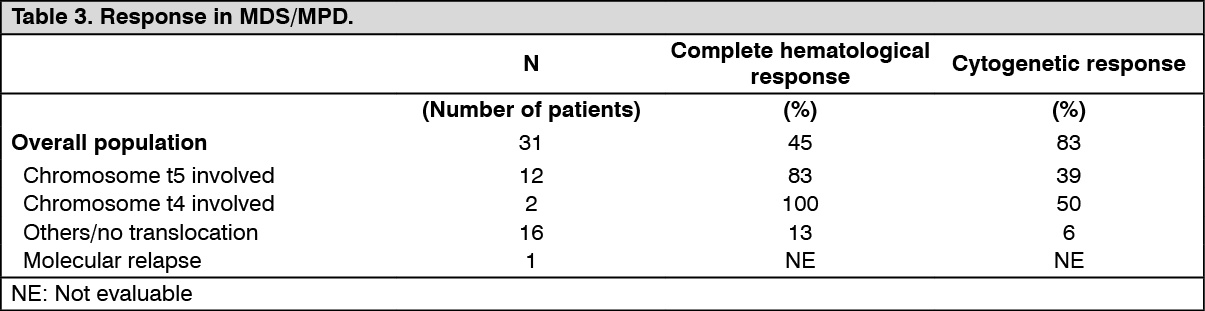 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image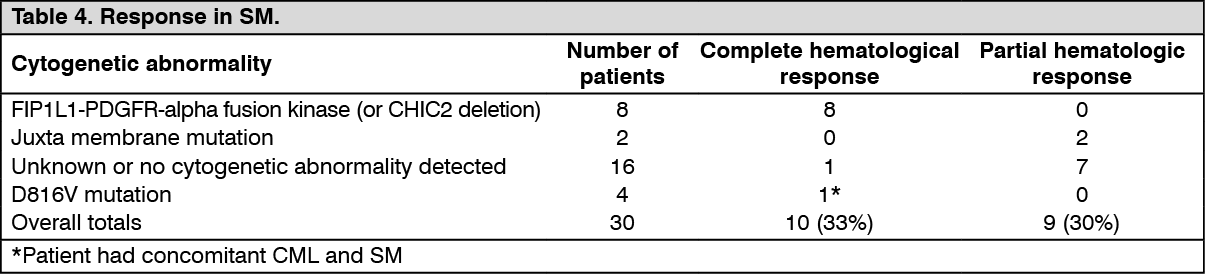 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image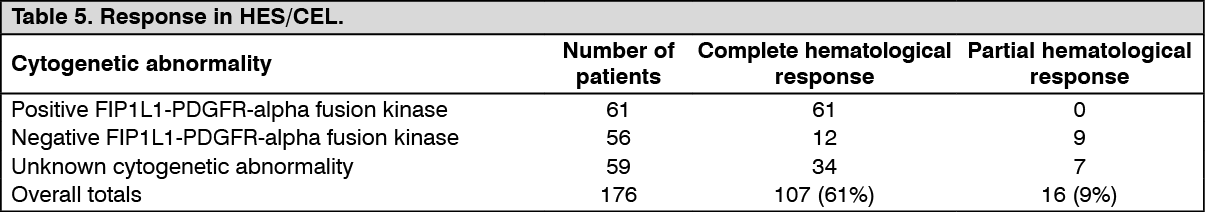 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image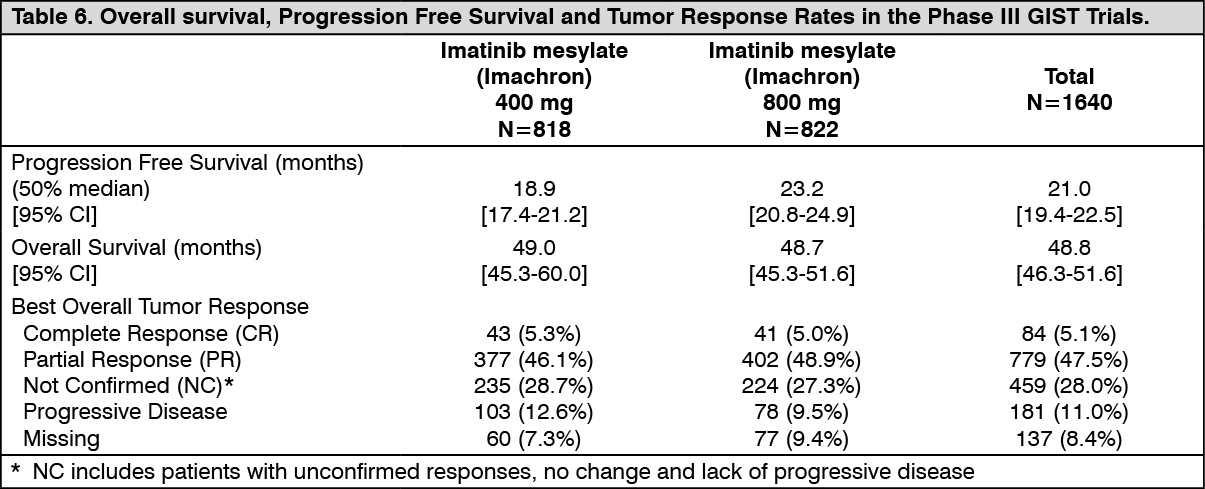 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image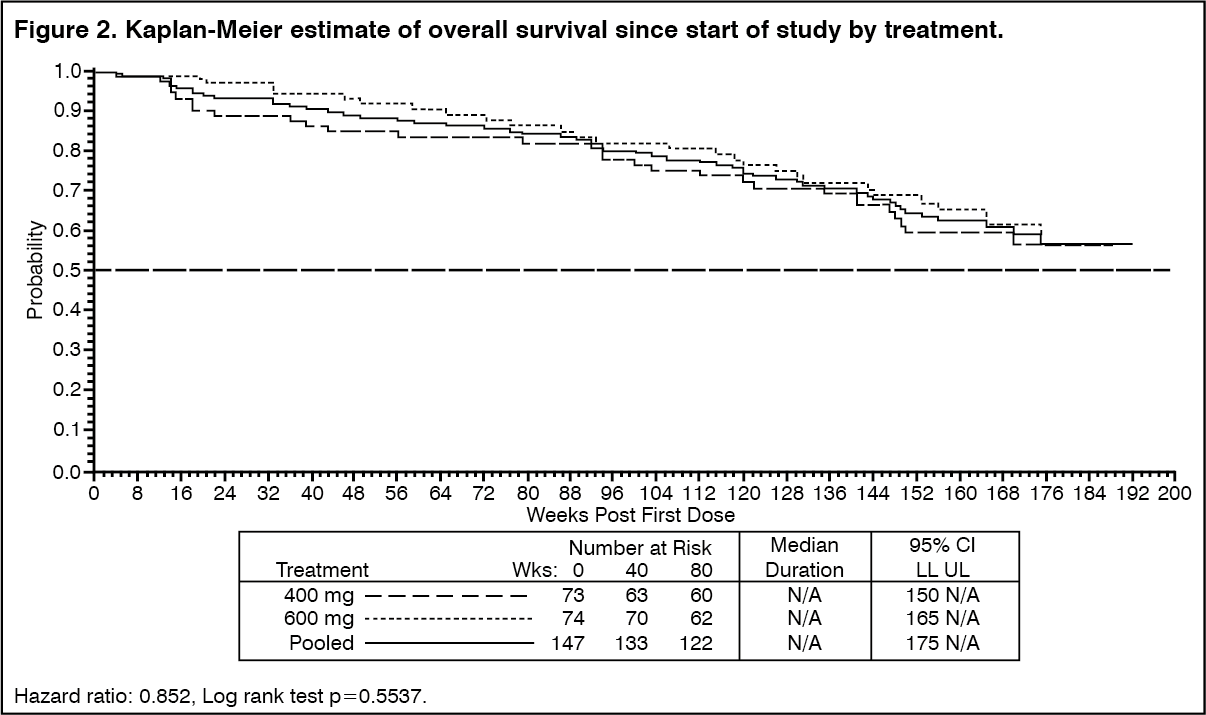 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image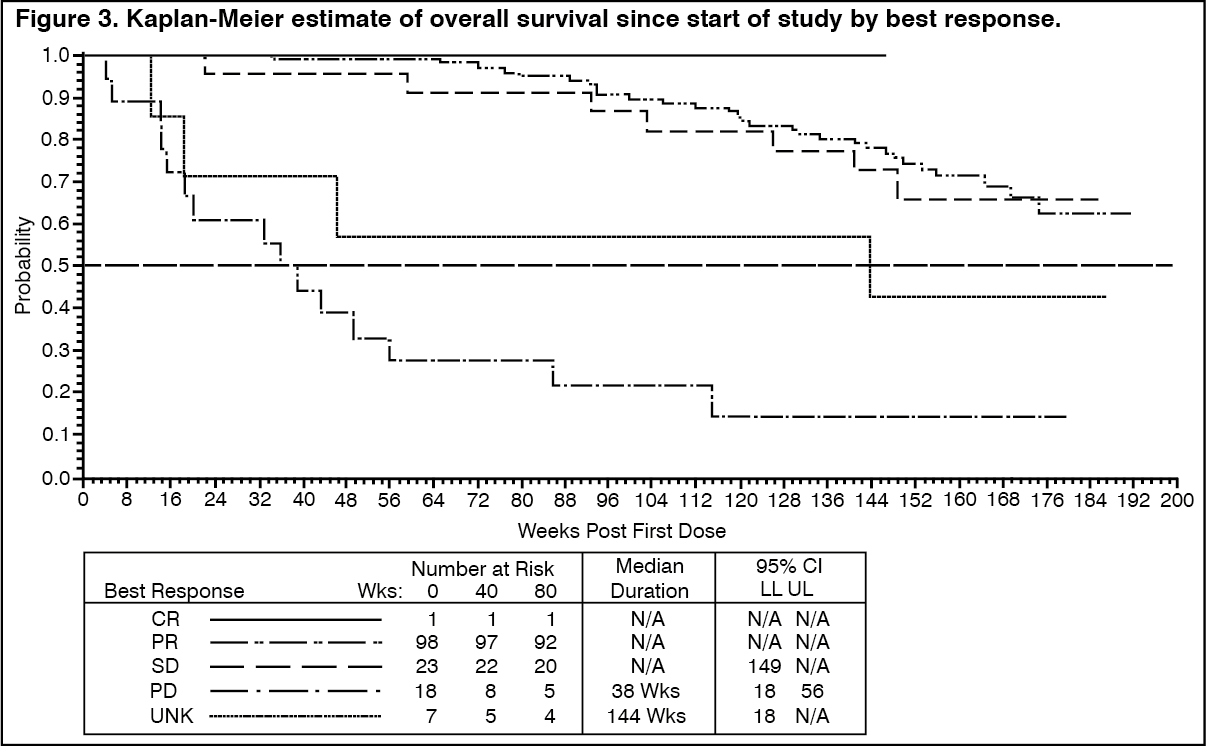 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image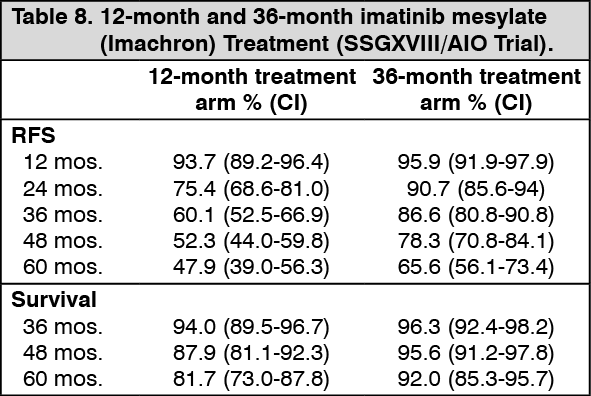 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image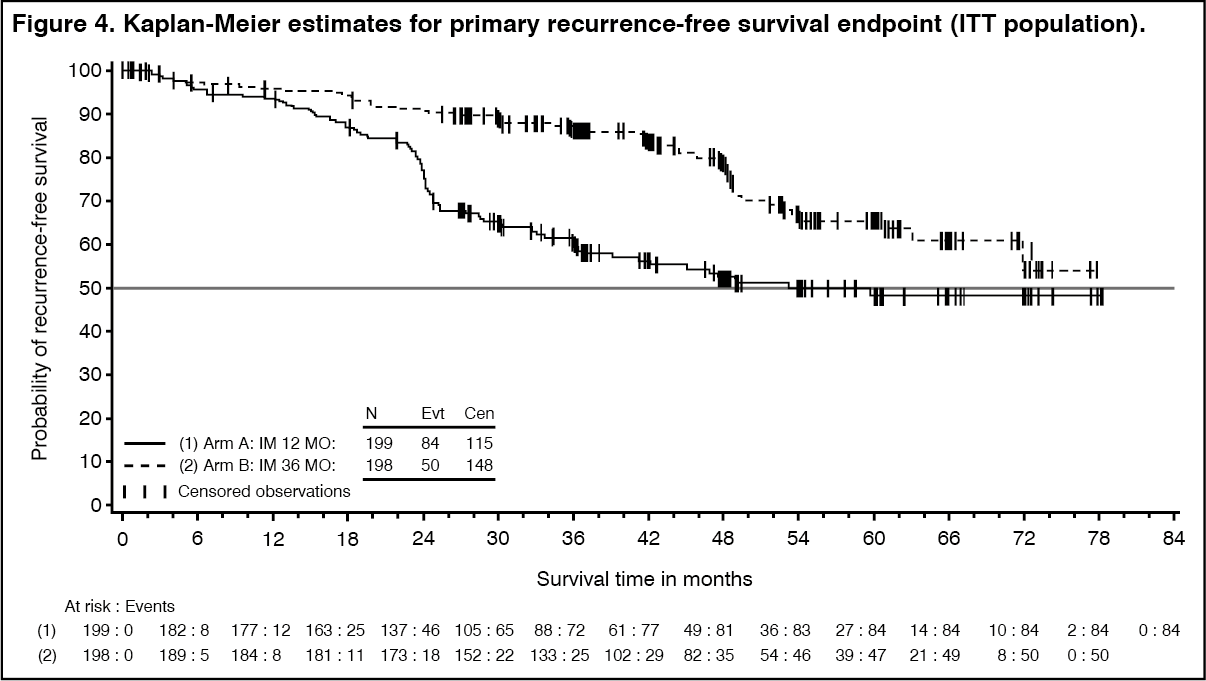 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image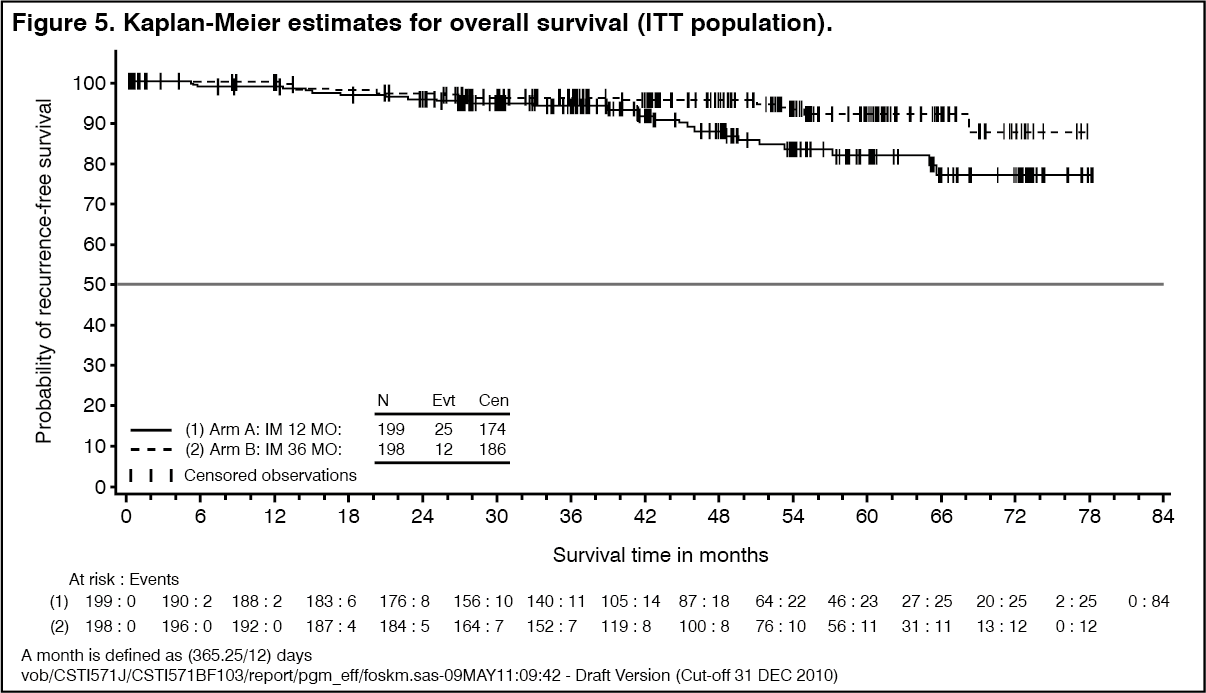 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image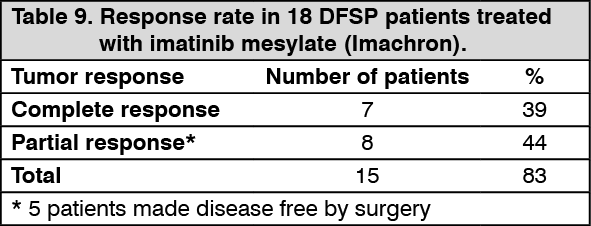 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image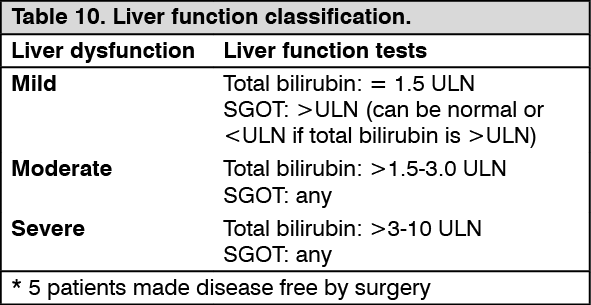 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image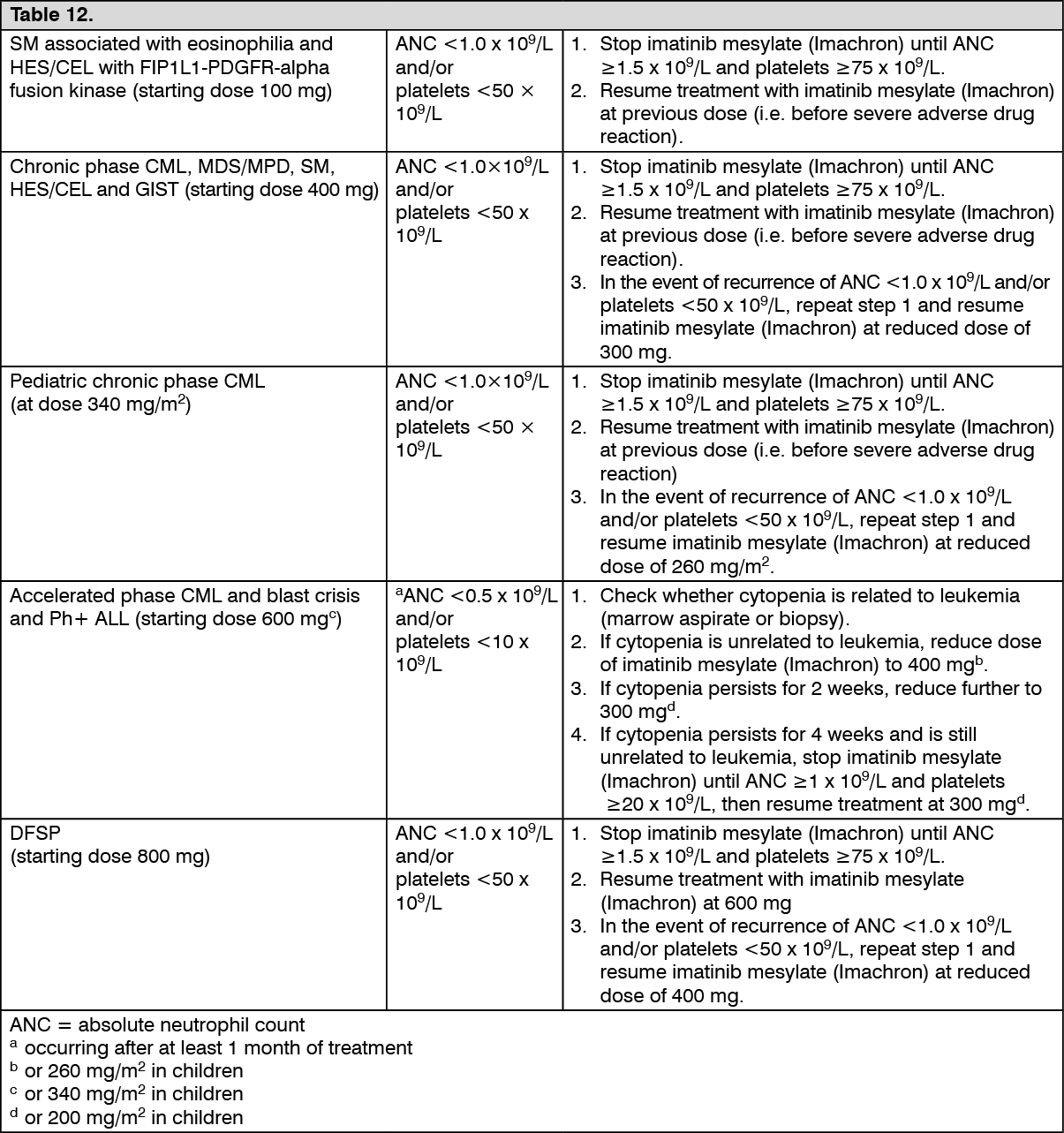 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image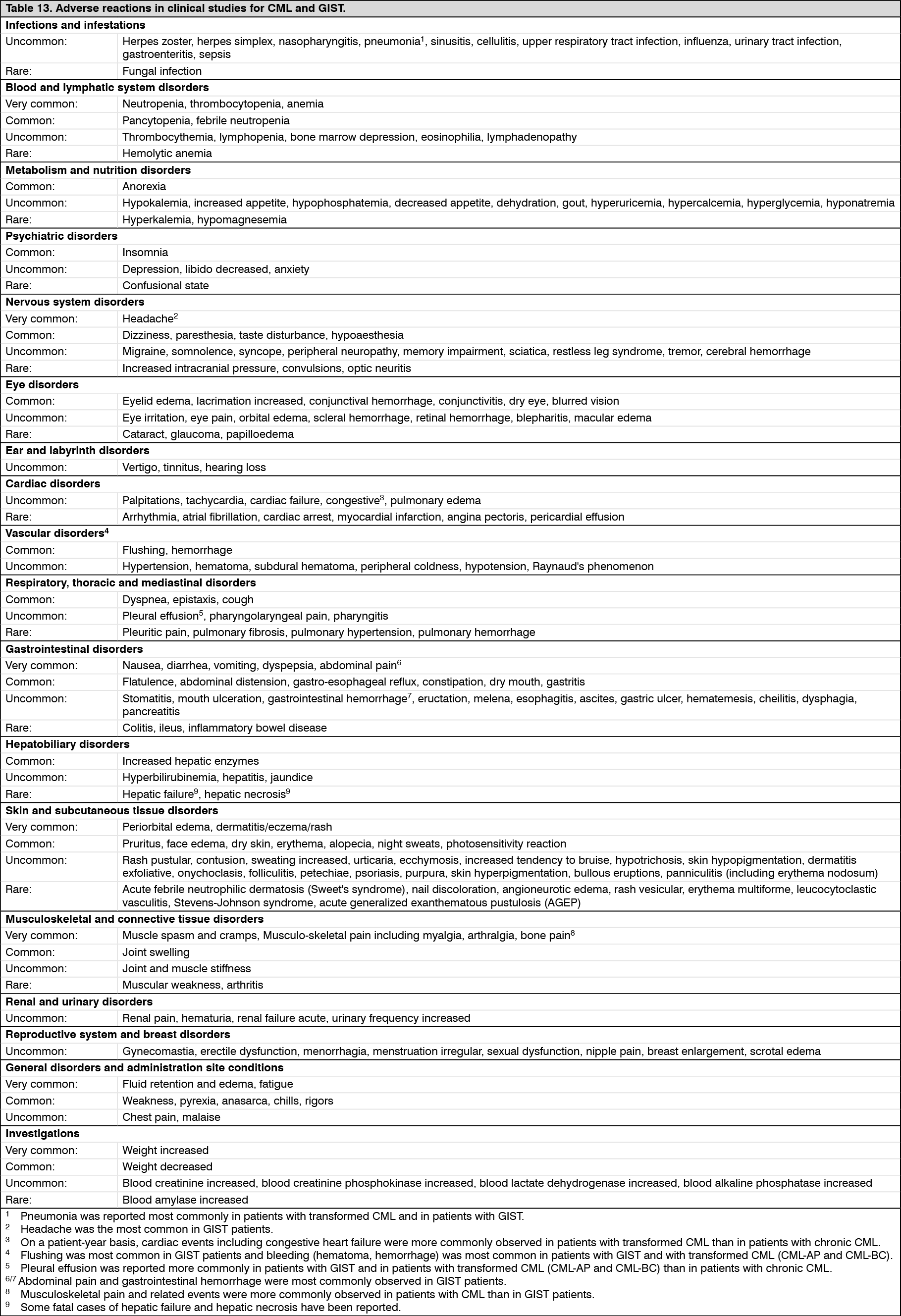 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out