



 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Montelukast: Pharmacotherapeutic group: Leukotriene receptor antagonist. ATC-code: R03D C03.
The cysteinyl leukotrienes (LTC4, LTD4, LTE4) are potent inflammatory eicosanoids released from various cells including mast cells and eosinophils. These important mediators bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in the pro-inflammatory cells (including eosinophils and certain myeloid stem cells). CysLTs have been correlated with the pathophysiology of asthma and allergic rhinitis. In allergic rhinitis, CysLTs are released from the nasal mucosa after allergen exposure during both early- and late-phase reactions and are associated with symptoms of allergic rhinitis. Intranasal challenge with CysLTs has been shown to increase nasal airway resistance and symptoms of nasal obstruction.
Montelukast is an orally active compound which binds with high affinity and selectivity to the CysLT1 receptor. Montelukast inhibits physiologic actions of LTD4 at the CysLT1 receptor without any agonist activity.
Levocetirizine: Pharmacotherapeutic group: antihistamine for systemic use, piperazine derivative. ATC code: R06A E09.
Levocetirizine, the (R) enantiomer of cetirizine, is a potent and selective antagonist of peripheral H1-receptors.
Binding studies revealed that levocetirizine has high affinity for human H1-receptors (Ki = 3.2 nmol/L). Levocetirizine has an affinity 2-fold higher than that of cetirizine (Ki = 6.3 nmol/L). Levocetirizine dissociates from H1-receptors with a half-life of 115 ± 38 min.
After single administration, levocetirizine shows a receptor occupancy of 90% at 4 hours and 57% at 24 hours.
Pharmacodynamic studies in healthy volunteers demonstrate that, at half the dose, levocetirizine has comparable activity to cetirizine, both in the skin and in the nose.
In a study comparing the effects of levocetirizine 5 mg, desloratadine 5 mg, and placebo on histamine-induced wheal and flare, levocetirizine treatment resulted in significantly decreased wheal and flare formation which was highest in the first 12 hours and lasted for 24 hours, (p<0.001) compared with placebo and desloratadine.
The onset of action of levocetirizine 5 mg in controlling pollen-induced symptoms has been observed at 1 hour post drug intake in placebo controlled trials in the model of the allergen challenge chamber.
In vitro studies (Boyden chambers and cell layers techniques) show that levocetirizine inhibits eotaxin-induced eosinophil transendothelial migration through both dermal and lung cells. A pharmacodynamic experimental study in vivo (skin chamber technique) showed three main inhibitory effects of levocetirizine 5 mg in the first 6 hours of pollen-induced reaction, compared with placebo in 14 adult patients: inhibition of VCAM-1 release, modulation of vascular permeability and a decrease in eosinophil recruitment.
ECGs did not show relevant effects of levocetirizine on QT interval.
Clinical Study: In a double-blind, randomized, comparative study of 14 days treatment duration the FDC of montelukast/levocetirizine hydrochloride 10/5 mg film-coated tablet was compared with Montelukast 10 mg tablet monotherapy and Levocetirizine hydrochloride 5 mg tablet monotherapy in the treatment of patients with seasonal allergic rhinitis.
A total of 279 subjects (mean age of 35.29 ± 11.58 years) were enrolled in the study and randomized to treatment with equal randomization (n=93) among three treatment arms. All subjects were Asian and 58% were male.
There was a statistically significant difference for the primary end point, mean change in the day time nasal symptom score (DTNSS) [composite score of rhinorrhoea, nasal congestion, nasal itching and sneezing] between the FDC and Montelukast (p<0.05) and Levocetirizine (p<0.05) for both PP and ITT populations. (See Table 1.)
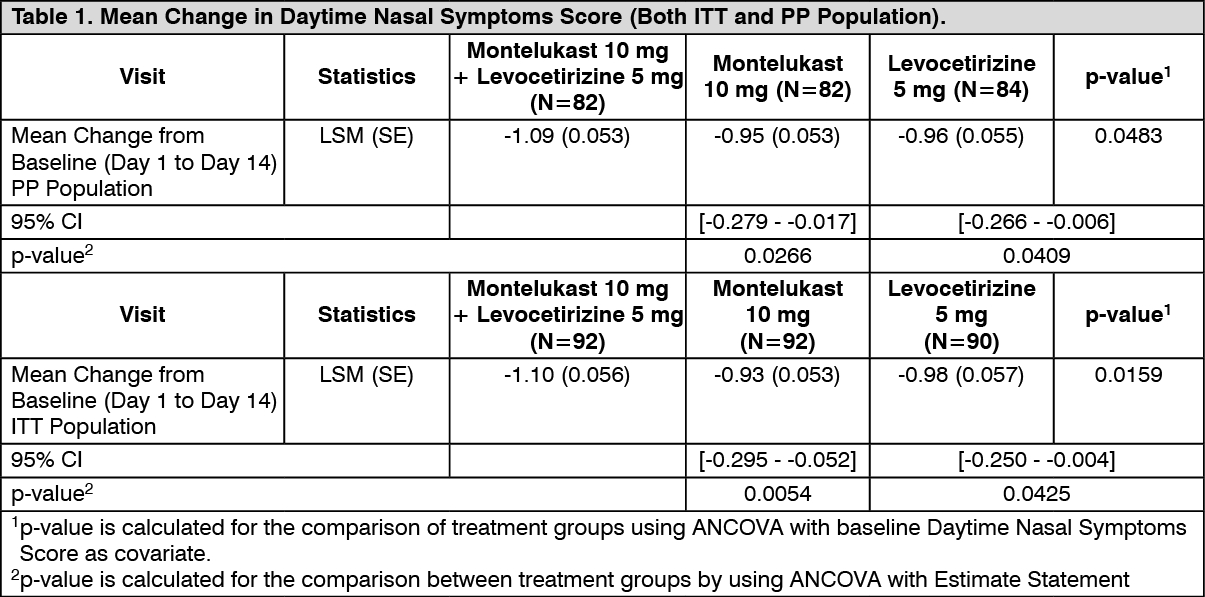 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOverall, the study indicated that FDC of Montelukast 10 mg and Levocetirizine hydrochloride 5 mg was safe and well tolerated; and safety profile is comparable to Montelukast 10 mg monotherapy and Levocetirizine hydrochloride 5 mg monotherapy.
Pharmacokinetics: No separate pharmacokinetic study was performed with the FDC of Montelukast and Levocetirizine to characterize its pharmacokinetics.
Absorption: Montelukast: Montelukast is rapidly absorbed following oral administration. For the 10 mg film-coated tablet, the mean peak plasma concentration (Cmax) is achieved 3 hours (Tmax) after administration in adults in the fasted state. The mean oral bioavailability is 64%. The oral bioavailability and Cmax are not influenced by a standard meal. Safety and efficacy were demonstrated in clinical trials where the 10 mg film-coated tablet was administered without regard to the timing of food ingestion.
Levocetirizine: The pharmacokinetics of levocetirizine are linear with dose- and time-independent with low inter-subject variability. The pharmacokinetic profile is the same when given as the single enantiomer or when given as cetirizine. No chiral inversion occurs during the process of absorption and elimination.
Levocetirizine is rapidly and extensively absorbed following oral administration. In adults, peak plasma concentrations are achieved 0.9 h after dosing. Steady state is achieved after two days. Peak concentrations are typically 270 ng/mL and 308 ng/mL following a single and a repeated 5 mg o.d. dose, respectively. The extent of absorption is dose-independent and is not altered by food, but the peak concentration is reduced and delayed.
Distribution: Montelukast: Montelukast is more than 99% bound to plasma proteins. The steady-state volume of distribution of montelukast averages 8-11 liters. Studies in rats with radio-labeled montelukast indicate minimal distribution across the blood-brain barrier. In addition, concentrations of radio-labeled material at 24 hours post-dose were minimal in all other tissues.
Levocetirizine: No tissue distribution data are available in humans, neither concerning the passage of levocetirizine through the blood-brain-barrier. In rats and dogs, the highest tissue levels are found in liver and kidneys, the lowest in the CNS compartment. Levocetirizine is 90% bound to plasma proteins. The distribution of levocetirizine is restrictive, as the volume of distribution is 0.4 L/kg.
Biotransformation: Montelukast: Montelukast is extensively metabolised. In studies with therapeutic doses, plasma concentrations of metabolites of montelukast are undetectable at steady state in adults and children.
Cytochrome P450 2C8 is the major enzyme in the metabolism of montelukast. Additionally CYP 3A4 and 2C9 may have a minor contribution, although itraconazole, an inhibitor of CYP 3A4, was shown not to change pharmacokinetic variables of montelukast in healthy subjects that received 10 mg montelukast daily. Based on in vitro results in human liver microsomes, therapeutic plasma concentrations of montelukast do not inhibit cytochromes P450 3A4, 2C9, 1A2, 2A6, 2C19, or 2D6. The contribution of metabolites to the therapeutic effect of montelukast is minimal.
Levocetirizine: The extent of metabolism of levocetirizine in humans is less than 14% of the dose and therefore differences resulting from genetic polymorphism or concomitant intake of enzyme inhibitors are expected to be negligible. Metabolic pathways include aromatic oxidation, N- and O-dealkylation and taurine conjugation. Dealkylation pathways are primarily mediated by CYP 3A4 while aromatic oxidation involved multiple and/or unidentified CYP isoforms. Levocetirizine had no effect on the activities of CYP isoenzymes 1A2, 2C9, 2C19, 2D6, 2E1 and 3A4 at concentrations well above peak concentrations achieved following a 5 mg oral dose. Due to its low metabolism and absence of metabolic inhibition potential, the interaction of levocetirizine with other substances, or vice-versa, is unlikely.
Elimination: Montelukast: The plasma clearance of montelukast averages 45 mL/min in healthy adults. Following an oral dose of radio-labeled montelukast, 86% of the radioactivity was recovered in 5-day faecal collections and <0.2% was recovered in urine. Coupled with estimates of montelukast oral bioavailability, this indicates that montelukast and its metabolites are excreted almost exclusively via the bile.
Levocetirizine: The plasma half-life in adults is 7.9 ± 1.9 hours. The mean apparent total body clearance is 0.63 mL/min/kg. The major route of excretion of levocetirizine and metabolites is via urine, accounting for a mean of 85.4% of the dose. Excretion via feces accounts for only 12.9% of the dose. Levocetirizine is excreted both by glomerular filtration and active tubular secretion.
Special Population: Studies in patients with renal and hepatic impairment have not been undertaken for FDC of Montelukast and Levocetirizine.
Renal impairment: Montelukast: Studies in patients with renal impairment have not been undertaken. Because montelukast and its metabolites are eliminated by the biliary route, no dose adjustment is anticipated to be necessary in patients with renal impairment.
Levocetirizine: The apparent body clearance of levocetirizine is correlated to the creatinine clearance. It is therefore recommended to adjust the dosing intervals of levocetirizine, based on creatinine clearance in patients with moderate and severe renal impairment. In anuric end stage renal disease subjects, the total body clearance is decreased by approximately 80% when compared to normal subjects. The amount of levocetirizine removed during a standard 4-hour hemodialysis procedure was <10%.
Hepatic impairment: Montelukast: No dosage adjustment is necessary for mild to moderate hepatic insufficiency. There are no data on the pharmacokinetics of montelukast in patients with severe hepatic insufficiency (Child-Pugh score >9).
Levocetirizine: Levocetirizine has not been studied in patients with hepatic impairment. The non-renal clearance (indicative of hepatic contribution) was found to constitute about 28% of the total body clearance in healthy adult subjects after oral administration. As levocetirizine is mainly excreted unchanged by the kidneys, it is unlikely that the clearance of levocetirizine is significantly decreased in patients with solely hepatic impairment.
The pharmacokinetics of levocetirizine in hepatically impaired subjects have not been tested. Patients with chronic liver diseases (hepatocellular, cholestatic, and biliary cirrhosis) given 10 or 20 mg of the racemic compound cetirizine as a single dose had a 50% increase in half-life along with a 40% decrease in clearance compared to healthy subjects.
Paediatric population: Levocetirizine: Data from a paediatric pharmacokinetic study with oral administration of a single dose of 5 mg levocetirizine in 14 children age 6 to 11 years with body weight ranging between 20 and 40 kg show that Cmax and AUC values are about 2-fold greater than that reported in healthy adult subjects in a cross-study comparison. The mean Cmax was 450 ng/mL, occurring at a mean time of 1.2 hours, weight-normalized, total body clearance was 30% greater, and the elimination half-life 24% shorter in this paediatric population than in adults. Dedicated pharmacokinetic studies have not been conducted in paediatric patients younger than 6 years of age. A retrospective population pharmacokinetic analysis was conducted in 324 subjects (181 children 1 to 5 years of age, 18 children 6 to 11 years of age, and 124 adults 18 to 55 years of age) who received single or multiple doses of levocetirizine ranging from 1.25 mg to 30 mg. Data generated from this analysis indicated that administration of 1.25 mg once daily to children 6 months to 5 years of age is expected to result in plasma concentrations similar to those of adults receiving 5 mg once daily.
Older people: Levocetirizine: Limited pharmacokinetic data are available in elderly subjects. Following once daily repeat oral administration of 30 mg levocetirizine for 6 days in 9 elderly subjects (6574 years of age), the total body clearance was approximately 33% lower compared to that in younger adults. The disposition of racemic cetirizine has been shown to be dependent on renal function rather than on age. This finding would also be applicable for levocetirizine, as levocetirizine and cetirizine are both predominantly excreted in urine. Therefore, the levocetirizine dose should be adjusted in accordance with renal function in elderly patients.
Gender: Levocetirizine: Pharmacokinetic results for 77 patients (40 men, 37 women) were evaluated for potential effect of gender. The half-life was slightly shorter in women (7.08 ± 1.72 hr) than in men (8.62 ± 1.84 hr); however, the body weight-adjusted oral clearance in women (0.67 ± 0.16 mL/min/kg) appears to be comparable to that in men (0.59 ± 0.12 mL/min/kg). The same daily doses and dosing intervals are applicable for men and women with normal renal function.
Race: Levocetirizine: The effect of race on levocetirizine has not been studied. As levocetirizine is primarily renally excreted, and there are no important racial differences in creatinine clearance, pharmacokinetic characteristics of levocetirizine are not expected to be different across races. No race-related differences in the kinetics of racemic cetirizine have been observed.
Toxicology: Preclinical safety data: Acute toxicity studies on rats and mice and repeated dose toxicity studies on rats were conducted of FDC of Montelukast and Levocetirizine. (See Table 2.)
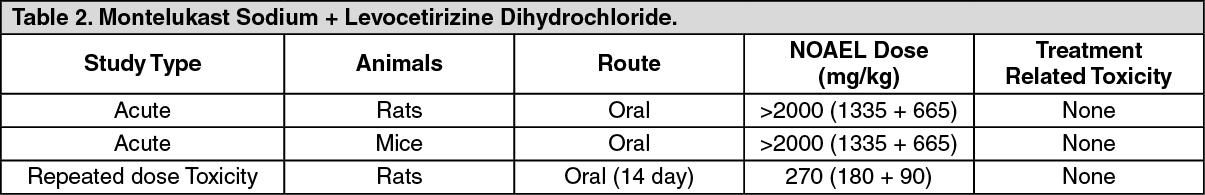 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe combination of montelukast and levocetirizine did not show any target organs of toxicity in 14-day oral (gavage) toxicity study in rats and the NOAEL established was 270 (180+90) mg/kg which is approximately 175 times (based on body surface area and considering human body weight as 60 kg) to the maximum recommended human daily dose [15 (10+5) mg/kg].
Carcinogenesis, Mutagenesis, Impairment of Fertility: No separate Carcinogenesis, Mutagenesis, Impairment of Fertility studies were performed with the FDC of Montelukast and Levocetirizine.
Montelukast: In animal toxicity studies, minor serum biochemical alterations in ALT, glucose, phosphorus and triglycerides were observed which were transient in nature. The signs of toxicity in animals were increased excretion of saliva, gastrointestinal symptoms, loose stools and ion imbalance. These occurred at dosages which provided >17-fold the systemic exposure seen at the clinical dosage. In monkeys, the adverse effects appeared at doses from 150 mg/kg/day (>232-fold the systemic exposure seen at the clinical dose). In animal studies, montelukast did not affect fertility or reproductive performance at systemic exposure exceeding the clinical systemic exposure by greater than 24-fold. A slight decrease in pup body weight was noted in the female fertility study in rats at 200 mg/kg/day (>69-fold the clinical systemic exposure). In studies in rabbits, a higher incidence of incomplete ossification, compared with concurrent control animals, was seen at systemic exposure >24-fold the clinical systemic exposure seen at the clinical dose. No abnormalities were seen in rats. Montelukast has been shown to cross the placental barrier and is excreted in breast milk of animals.
No deaths occurred following a single oral administration of montelukast sodium at doses up to 5000 mg/kg in mice and rats (15,000 mg/m2 and 30,000 mg/m2 in mice and rats, respectively), the maximum dose tested. This dose is equivalent to 25,000 times the recommended daily adult human dose (based on an adult patient weight of 50 kg).
Montelukast was determined not to be phototoxic in mice for UVA, UVB or visible light spectra at doses up to 500 mg/kg/day (approximately >200-fold based on systemic exposure).
Montelukast was neither mutagenic in in vitro and in vivo tests nor tumorigenic in rodent species.
No evidence of tumorigenicity was seen in carcinogenicity studies of either 2 years in Sprague-Dawley rats or 92 weeks in mice at oral gavage doses up to 200 mg/kg/day or 100 mg/kg/day, respectively. The estimated exposure in rats was approximately 120 and 75 times the AUC for adults and children, respectively, at the maximum recommended daily oral dose. The estimated exposure in mice was approximately 45 and 25 times the AUC for adults and children, respectively, at the maximum recommended daily oral dose.
Montelukast demonstrated no evidence of mutagenic or clastogenic activity in the following assays: the microbial mutagenesis assay, the V-79 mammalian cell mutagenesis assay, the alkaline elution assay in rat hepatocytes, the chromosomal aberration assay in Chinese hamster ovary cells, and in the in vivo mouse bone marrow chromosomal aberration assay.
In fertility studies in female rats, montelukast produced reductions in fertility and fecundity indices at an oral dose of 200 mg/kg (estimated exposure was approximately 70 times the AUC for adults at the maximum recommended daily oral dose). No effects on female fertility or fecundity were observed at an oral dose of 100 mg/kg (estimated exposure was approximately 20 times the AUC for adults at the maximum recommended daily oral dose). Montelukast had no effects on fertility in male rats at oral doses up to 800 mg/kg (estimated exposure was approximately 160 times the AUC for adults at the maximum recommended daily oral dose).
Levocetirizine: No carcinogenicity studies have been performed with levocetirizine. However, evaluation of cetirizine carcinogenicity studies is relevant for determination of the carcinogenic potential of levocetirizine. In a 2-year carcinogenicity study, in rats, cetirizine was not carcinogenic at dietary doses up to 20 mg/kg (approximately 15 times the maximum recommended daily oral dose in adults, approximately 10 times the maximum recommended daily oral dose in children 6 to 11 years of age and approximately 15 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). In a 2-year carcinogenicity study in mice, cetirizine caused an increased incidence of benign hepatic tumors in males at a dietary dose of 16 mg/kg (approximately 6 times the maximum recommended daily oral dose in adults, approximately 4 times the maximum recommended daily oral dose in children 6 to 11 years of age, and approximately 6 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). No increased incidence of benign tumors was observed at a dietary dose of 4 mg/kg (approximately 2 times the maximum recommended daily oral dose in adults, equivalent to the maximum recommended daily oral dose in children 6 to 11 years of age and approximately 2 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). The clinical significance of these findings during long-term use of levocetirizine is not known.
Levocetirizine was not mutagenic in the Ames test, and not clastogenic in the human lymphocyte assay, the mouse lymphoma assay, and in vivo micronucleus test in mice.
In a fertility and general reproductive performance study in mice, cetirizine did not impair fertility at an oral dose of 64 mg/kg (approximately 25 times the recommended daily oral dose in adults on mg/m2 basis).
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction.