



 Sign Out
Sign Out
Pharmacodynamics: Platelets: In clinical trials using normal volunteers, Celecoxib at single doses up to 800 mg and multiple doses of 600 mg twice daily for up to 7 days duration (higher than recommended therapeutic doses) had no effect on reduction of platelet aggregation or increase in bleeding time. Because of its lack of platelet effects, Celecoxib is not a substitute for Aspirin for cardiovascular prophylaxis. It is not known if there are any effects of Celecoxib on platelets that may contribute to the increased risk of serious cardiovascular thrombotic adverse events associated with the use of Celecoxib.
Fluid Retention: Inhibition of PGE2 synthesis may lead to sodium and water retention through increased reabsorption in the renal medullary thick ascending loop of Henle and perhaps other segments of the distal nephron. In the collecting ducts, PGE2 appears to inhibit water reabsorption by counteracting the action of antidiuretic hormone.
Pharmacokinetics: Absorption: Peak plasma levels of Celecoxib occur approximately 3 hrs after an oral dose. Under fasting conditions, both peak plasma levels (Cmax) and area under the curve (AUC) are roughly dose-proportional up to 200 mg BID; at higher doses there are less than proportional increases in Cmax and AUC. Absolute bioavailability studies have not been conducted. With multiple dosing, steady-state conditions are reached on or before Day 5. The pharmacokinetic parameters of Celecoxib in a group of healthy subjects are shown in Table 1. (See Table 1.)
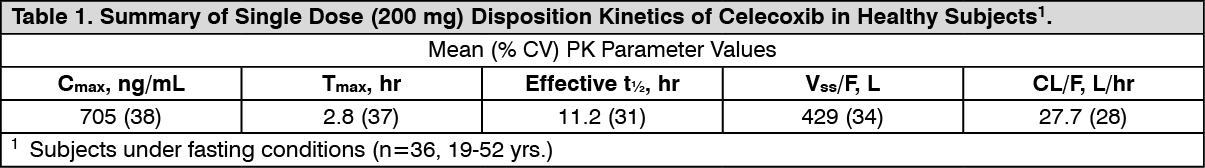 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFood Effects: When Celecoxib capsules were taken with a high fat meal, peak plasma levels were delayed for about 1 to 2 hours with an increase in total absorption (AUC) of 10% to 20%. Under fasting conditions, at doses above 200 mg, there is less than a proportional increase in Cmax and AUC, which is thought to be due to the low solubility of the drug in aqueous media.
Co-administration of Celecoxib with an aluminum- and magnesium-containing antacids resulted in a reduction in plasma Celecoxib concentrations with a decrease of 37% in Cmax and 10% in AUC. Celecoxib, at doses up to 200 mg twice daily, can be administered without regard to timing of meals. Higher doses (400 mg twice daily) should be administered with food to improve absorption. In healthy adult volunteers, the overall systemic exposure (AUC) of Celecoxib was equivalent when Celecoxib was administered as intact capsule or capsule contents sprinkled on apple sauce. There were no significant alterations in Cmax, Tmax or t1/2 after administration of capsule contents on applesauce.
Distribution: In healthy subjects, Celecoxib is highly protein bound (~97%) within the clinical dose range. In vitro studies indicate that Celecoxib binds primarily to albumin and, to a lesser extent, α1-acid glycoprotein. The apparent volume of distribution at steady state (Vss/F) is approximately 400 L, suggesting extensive distribution into the tissues. Celecoxib is not preferentially bound to red blood cells.
Metabolism: Celecoxib metabolism is primarily mediated via CYP2C9. Three metabolites, a primary alcohol, the corresponding carboxylic acid and its glucuronide conjugate, have been identified in human plasma. These metabolites are inactive as COX-1 or COX-2 inhibitors.
CYP2C9 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity, such as those homozygous for the CYP2C9*2 and CYP2C9*3 polymorphisms. Limited data from 4 published reports that included a total of 8 subjects with the homozygous CYP2C9*3/*3 genotype showed Celecoxib systemic levels that were 3- to 7-fold higher in these subjects compared to subjects with CYP2C9*1/*1 or *I/*3 genotypes. The pharmacokinetics of Celecoxib have not been evaluated in subjects with other CYP2C9 polymorphisms, such as *2, *5, *6, *9 and *11. It is estimated that the frequency of the homozygous *3/*3 genotype is 0.3% to 1.0% in various ethnic groups.
Excretion: Celecoxib is eliminated predominantly by hepatic metabolism with little (<3%) unchanged drug recovered in the urine and feces. Following a single oral dose of radiolabeled drug, approximately 57% of the dose was excreted in the feces and 27% was excreted into the urine. The primary metabolite in both urine and feces was the carboxylic acid metabolite (73% of dose) with low amounts of the glucuronide also appearing in the urine. It appears that the low solubility of the drug prolongs the absorption process making terminal half-life (t1/2) determinations more variable. The effective half-life is approximately 11 hours under fasted conditions. The apparent plasma clearance (CL/F) is about 500 mL/min.
Geriatric: At steady state, elderly subjects (over 65 years old) had a 40% higher Cmax and a 50% higher AUC compared to the young subjects. In elderly females, Celecoxib Cmax and AUC are higher than those for elderly males, but these increases are predominantly due to lower body weight in elderly females. Dose adjustment in the elderly is not generally necessary. However, for patients of less than 50 kg in body weight, initiate therapy at the lowest recommended dose.
Pediatric: The steady state pharmacokinetics of Celecoxib administered as an investigational oral suspension was evaluated in 152 JRA patients 2 years to 17 years of age weighing ≥10 kg with pauciarticular or polyarticular course JRA and in patients with systemic onset JRA. Population pharmacokinetic analysis indicated that the oral clearance (unadjusted for body weight) of Celecoxib increases less than proportionally to increasing weight, with 10 kg and 25 kg patients predicted to have 40% and 24% lower clearance, respectively, compared with a 70 kg adult RA patient.
Twice-daily administration of 50 mg capsules to JRA patients weighing ≥12 to ≤25 kg and 100 mg capsules to JRA patients weighing >25 kg should achieve plasma concentrations similar to those observed in a clinical trial that demonstrated the non-inferiority of Celecoxib to naproxen 7.5 mg/kg twice daily (see Dosage & Administration). Celecoxib has not been studied in JRA patients under the age of 2 years, in patients with body weight less than 10 kg (22 lbs), or beyond 24 weeks.
Race: Meta-analysis of pharmacokinetic studies has suggested an approximately 40% higher AUC of Celecoxib in Blacks compared to Caucasians. The cause and clinical significance of this finding is unknown.
Hepatic Insufficiency: A pharmacokinetic study in subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment has shown that steady-state Celecoxib AUC is increased about 40% and 180%, respectively, above that seen in healthy control subjects. Therefore, the daily recommended dose of Celecoxib capsules should be reduced by approximately 50% in patients with moderate (Child-Pugh Class B) hepatic impairment. Patients with severe hepatic impairment (Child-Pugh Class C) have not been studied. The use of Celecoxib in patients with severe hepatic impairment is not recommended.
Renal Insufficiency: In a cross-study comparison, Celecoxib AUC was approximately 40% lower in patients with chronic renal insufficiency (GFR 35-60 mL/min) than that seen in subjects with normal renal function. No significant relationship was found between GFR and Celecoxib clearance.
Patients with severe renal insufficiency have not been studied. Similar to other NSAIDs, Celecoxib is not recommended in patients with severe renal insufficiency.
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Celecoxib was not carcinogenic in rats given oral doses up to 200 mg/kg for males and 10 mg/kg for females (approximately 2- to 4-fold the human exposure as measured by the AUC0-24 at 200 mg twice daily) or in mice given oral doses up to 25 mg/kg for males and 50 mg/kg for females (approximately equal to human exposure as measured by the AUC0-24 at 200 mg twice daily) for two years.
Celecoxib was not mutagenic in an Ames test and a mutation assay in Chinese hamster ovary (CHO) cells, nor clastogenic in a chromosome aberration assay in CHO cells and an in vivo micronucleus test in rat bone marrow.
Celecoxib did not impair male and female fertility in rats at oral doses up to 600 mg/kg/day (approximately 11-fold human exposure at 200 mg twice daily based on the AUC0-24).
Animal Toxicology: An increase in the incidence of background findings of spermatocele with or without secondary changes such as epididymal hypospermia as well as minimal to slight dilation of the seminiferous tubules was seen in the juvenile rat. These reproductive findings while apparently treatment-related did not increase in incidence or severity with dose and may indicate an exacerbation of a spontaneous condition. Similar reproductive findings were not observed in studies of juvenile or adult dogs or in adult rats treated with Celecoxib. The clinical significance of this observation is unknown.