Monotherapy: Colon, colorectal and breast cancer: Given as monotherapy, the recommended starting dose for Capecitabine in the adjuvant treatment of colon cancer, in the treatment of metastatic colorectal cancer or of locally advanced or metastatic breast cancer is 1,250 mg/m
2 administered twice daily (morning and evening; equivalent to 2,500 mg/m
2 total daily dose) for 14 days followed by a 7-day rest period. Adjuvant treatment in patients with stage III colon cancer is recommended for a total of 6 months.
Combination therapy: Colon, colorectal and gastric cancer: In combination treatment, the recommended starting dose of Capecitabine should be reduced to 800-1,000 mg/m
2 when administered twice daily for 14 days followed by a 7-day rest period or to 625 mg/m
2 twice daily when administered continuously.
Breast cancer: In combination with docetaxel, the recommended starting dose of Capecitabine in the treatment of metastatic breast cancer is 1,250 mg/m
2 twice daily for 14 days followed by a 7-day rest period, combined with docetaxel at 75 mg/m
2 as a 1 hour intravenous infusion every 3 weeks. Pre-medication with an oral corticosteroid such as dexamethasone should be started prior to docetaxel administration for patients receiving the Capecitabine plus docetaxel combination.
Or as prescribed by the physician.
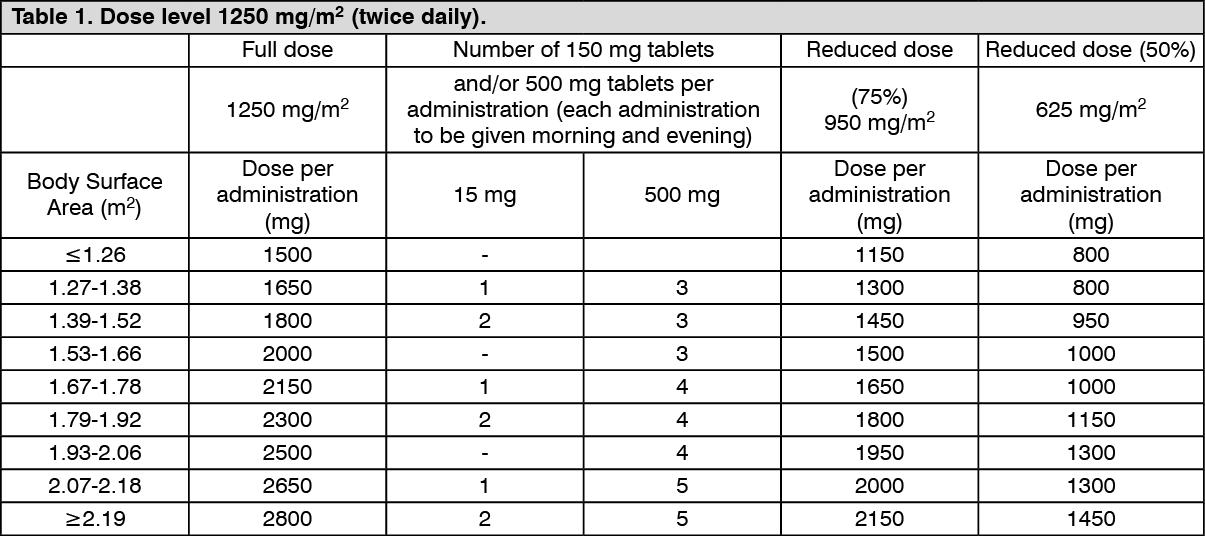
Standard and reduced dose calculations according to body surface area for a starting dose of Capecitabine of 1,250 mg/m
2 (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Standard and reduced dose calculations according to body surface area for a starting dose of Capecitabine of 1,000 mg/m
2 (see Table 2).
Click on icon to see table/diagram/image
Dose adjustments during treatment: General: Toxicity due to Capecitabine administration may be managed by symptomatic treatment and/or modification of the dose (treatment interruption or dose reduction). Once the dose has been reduced, it should not be increased at a later time. For those toxicities considered by the treating physician to be unlikely to become serious or life-threatening, e.g. alopecia, altered taste, nail changes, treatment can be continued at the same dose without reduction or interruption. Doses of Capecitabine omitted for toxicity are not replaced. The following are the recommended dose modifications for toxicity: (see Table 3).
Click on icon to see table/diagram/image
Haematology: Patients with baseline neutrophil counts of <1.5 x 10
9/L and/or thrombocyte counts of <100 x 10
9/L should not be treated with Capecitabine. If unscheduled laboratory assessments during a treatment cycle show that the neutrophil count drops below 1.0 x 10
9/L or that the platelet count drops below 75 x 10
9/L, treatment with Capecitabine should be interrupted.
Posology adjustments for special populations: Hepatic impairment: Insufficient safety and efficacy data are available in patients with hepatic impairment to provide a dose adjustment recommendation. No information is available on hepatic impairment due to cirrhosis or hepatitis.
Renal impairment: Capecitabine is contraindicated in patients with severe renal impairment (creatinine clearance below 30 mL/min [Cockcroft and Gault] at baseline).
Elderly: During Capecitabine monotherapy, no adjustment of the starting dose is needed. However, grade 3 or 4 treatment-related adverse reactions were more frequent in patients >60 years of age compared to younger patients.
Pediatric population: There is no relevant use of Capecitabine in the pediatric population in the indications for colon, colorectal, gastric and breast cancer.
Method of administration: Capecitabine tablets should be swallowed with water within 30 minutes after a meal.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image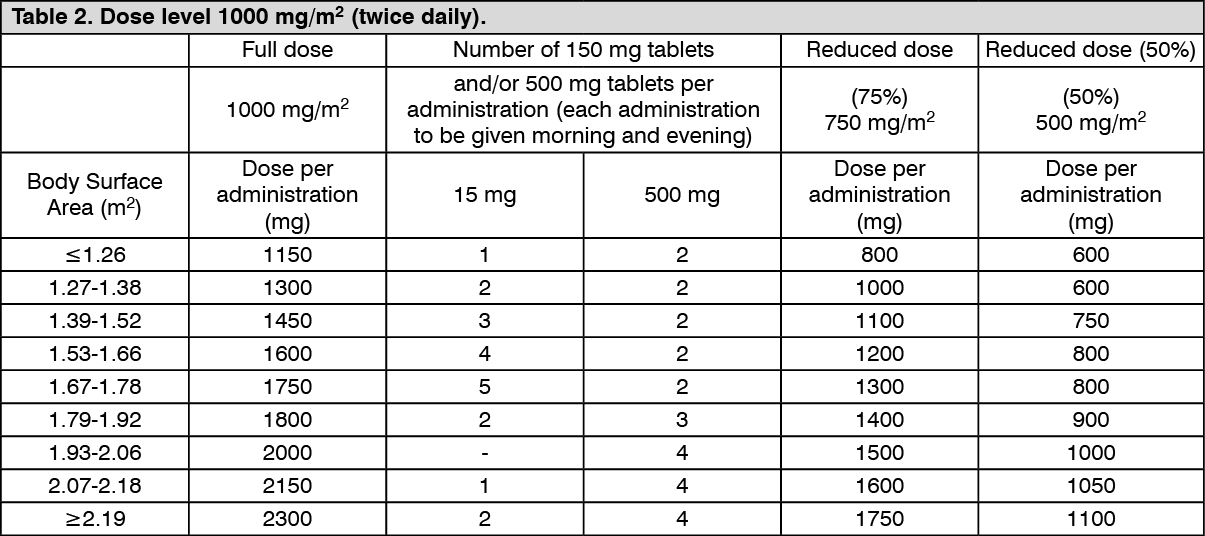 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image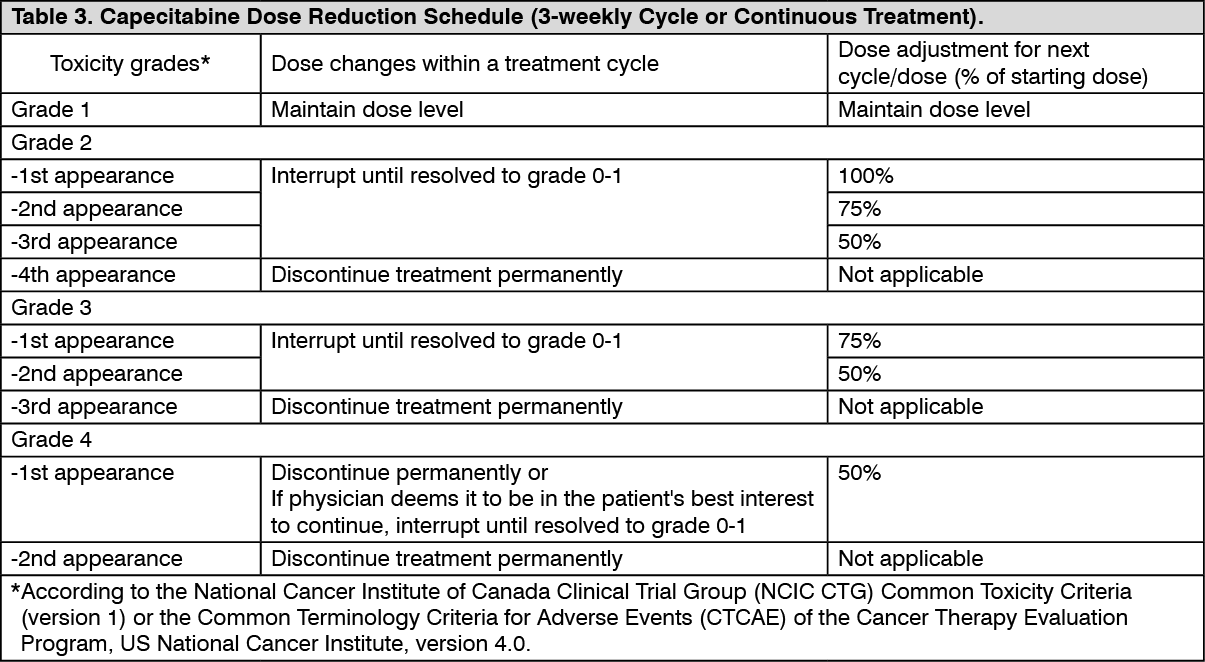 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image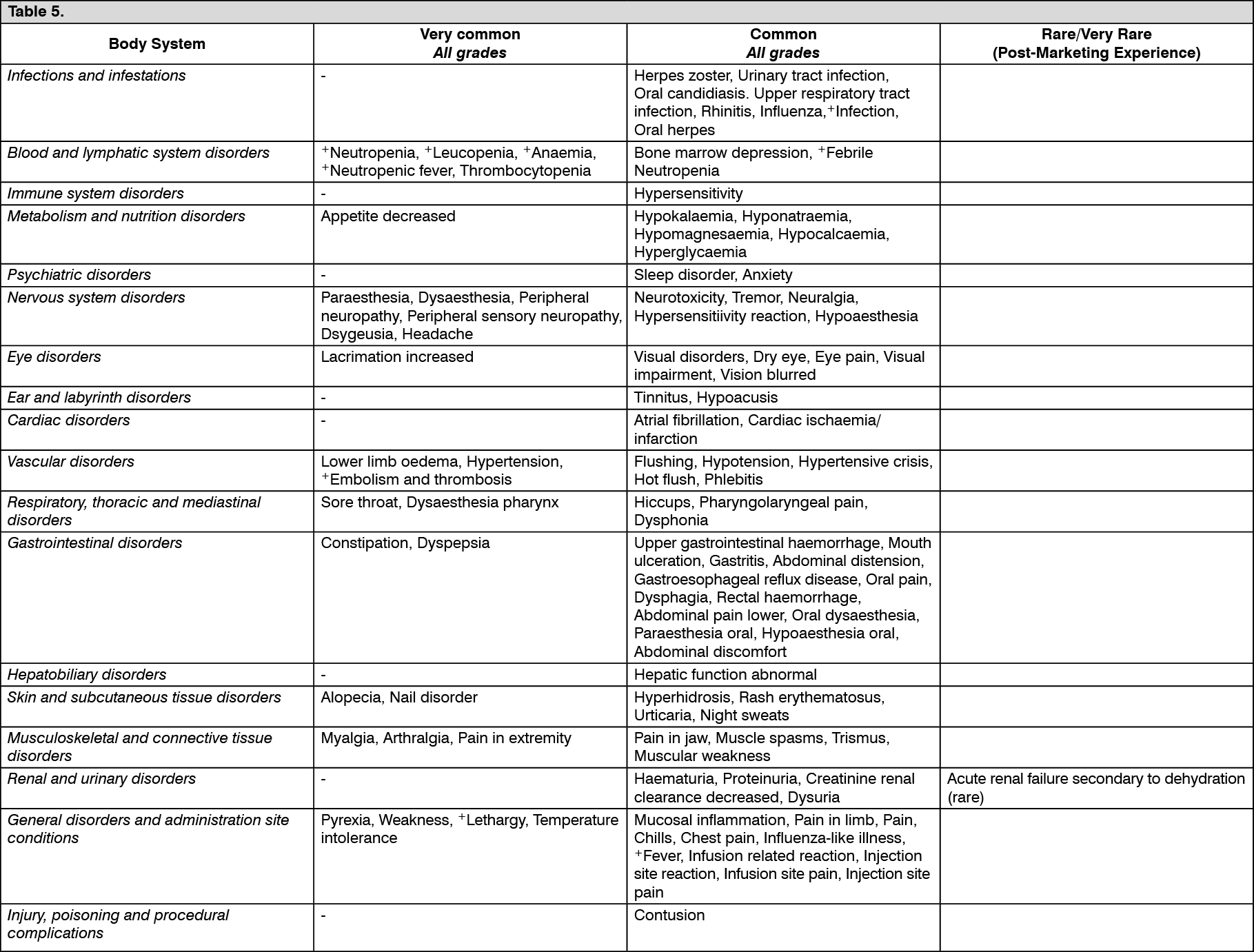 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out